Evidence that PrP GPI signal sequence matters
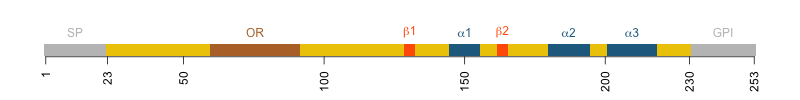
Diagram of human PrP. Amino acids 23-230 are the mature protein. 231-253 are the GPI signal.
Human PrP is 253 codons long, but there are only 208 amino acids in the mature, post-translationally modified protein. The first 22 residues are a signal peptide (SP) that directs PrP to the ER and ultimately the cell surface. The last 23 residues are a GPI signal that gets chopped off and replaced with a GPI anchor that is inserted into the ER membrane and ultimately the outer leaflet of the cell membrane. That which isn’t there can’t matter, right? If the 23 amino acids of the GPI signal are not even present in the mature protein, how can they affect the development of prion disease? And yet a fair amount of evidence now indicates that they do matter. This blog post will unpack what we know.
background
GPI stands for glycosylphosphatidylinositol. It’s a sugar-and-fat chain that gets attached to the C terminus of some proteins and anchors them into the membrane. I took a deeper dive on GPI anchors in this post if you want more background. That PrP is GPI-anchored was shown empirically in a series of papers from the Prusiner lab [Stahl 1987, Stahl 1990, Stahl 1992]. GPI anchors are not just one thing — there can be a variety of compositions. Through a series of enzymatic digestions and mass spectrometry experiments, they identified at least 6 GPI anchor compositions associated with PrP, comprising different combinations of mannose, galactose, GalNAc, and sialic acid, sitting on top of the phosphatidylinositol [Stahl 1992].
evidence that the GPI anchor matters
First things first, the presence vs. absence of the GPI anchor matters a lot. As reviewed in this post, a series of mouse models have shown that expression of “anchorless” PrP causes an atypical prion disease (sometimes called PrP cerebral amyloid angiopathy) characterized by intense amyloid deposition including onto blood vessels [Chesebro 2005, Trifilo 2008, Chesebro 2010, Stohr 2011]. This is mirrored by a rare subset of genetic prion disease in humans, where late truncating mutations that remove the GPI anchor, leading to secreted PrP, cause amyloid deposition in peripheral tissues as well as the brain, leading to inflammatory bowel disease-like symptoms, progressing to peripheral neuropathy and then slowly progressive dementia over 20 years [Mead & Reilly 2015]. The secreted PrP in these anchorless mouse models is (almost) entirely lacking in N-linked glycans at residues N181 and N197.
But of course the presence vs. absence of the GPI anchor matters, since you are talking about a difference in whether PrP is on the cell surface or secreted into extracellular space. What about the the amino acid sequence of the GPI signal and the composition of the GPI anchor itself?
The evidence from human genetics comes from M232R, a genetic variant that affects only the GPI signal and appears to increase the risk of prion disease by about 5-fold [Nozaki 2010, Minikel 2016]. But how does it do that?
One study from Tetsuyuki Kitamoto’s lab expressed every possible mutant at codons 228, 230, 231, and 232 in N2a cells and tested their GPI anchoring efficiency and ability to convert to PrPSc [Hizume 2010]. That study reported that 232R PrP was under-expressed on the cell surface, and that more of the 232R PrP was secreted into cell culture media. Whereas WT PrP was only released when cells were treted with PI-PLC — an enzyme that cleaves GPI anchors — it is shown in Figure 2C that some 232R PrP is either shed or secreted from the cells even in the absence of PI-PLC. They used Triton X-114 phase partitioning [Bordier 1981], a procedure which exploits the temperature-dependent solubility of the detergent Triton X-114. This detergent can dissolve at low temperatures, but at high temperatures it separates into an aqueous phase and a detergent phase. Membrane proteins and membrane-anchored (e.g. GPI) proteins will be in the detergent phase, while secreted proteins will be in the aqueous phase. Whereas WT PrP was found about equally in both, 232R PrP was found predominantly in the aqueous phase, suggesting more of it was not GPI-anchored. But an enormous caveat to all of this is that the system used was overexpression in transfected N2a cells, which may or may not reflect the real situation in vivo. And indeed, later work by this same group contradicted some of these findings (see below).
There have been various other cell culture studies, including one in primary neurons from knockout mice [Bate 2016]. PrPCpurified from N9 glial cells was added to cell culture media and they watched if and where the PrP was taken up by the knockout neurons. They found that PrP with its GPI anchor intact was taken up and localized to synapses, whereas if they used enzymes to remove either the sialic acid or one of the acyl chain from the GPI anchor, the synaptic localization was lost. Although it was an artificial system, this study provided at least some evidence that a change in GPI anchor composition could lead to a change in PrP’s behavior, in this case, its localization.
Perhaps the first in vivo evidence that changes in the GPI signal can affect prion disease came from a study replacing PrP’s GPI signal with that of Thy1, a different GPI-anchored protein [Puig 2019]. Thy1’s GPI anchor, unlike PrP’s, does not contain sialic acid, so they hypothesized that as was shown in the primary neurons [Bate 2016], an unsialyated GPI anchor would lead PrP to behave differently. They created a transgenic mouse expressing PrP with the Thy1 GPI anchor. By Western blot, the transgenic mice appeared to express about the same amount of PrP as wild-type mice, or if anything, slightly more. And yet the incubation time when inoculated with prions was longer than wild-type mice: 195 vs. 155 dpi for RML, 156 vs. 144 dpi for 22L. Although the authors concluded that “the delay was independent of prion strain”, and true, the direction of effect was the same, it actually looks to me like there may be some difference in the magnitude of effect caused by the Thy1 GPI anchor (+26% for RML vs. +8% for 22L), which may be interesting in light of later results (see below). They also reported several other functional differences in the PrP-Thy1GPI mice, including changed localization, different neuropathology after prion infection, and a striking reduction in the amount of PrP shedding (cleavage at the GPI anchor, releasing PrP into extracellular space).
Last year, a new in vivo study from the Kitamoto group, this one using human prions in transgenic mice, provided more evidence for how the M232R variant in particular can influence prion disease [Kobayashi 2023]. They made knock-in mice expressing either wild-type (232M) human PrP, or 232R human PrP, and they performed detailed biochemical studies on these in addition to prion infection experiments. They found that 232R PrP was indeed GPI-anchored and in a Triton X-114 partitioning assay, it partitioned to the detergent side (where GPI-anchored proteins go) just as much as wild-type PrP did. This refuted the finding in N2a cells from [Hizume 2010] that 232R PrP is not properly anchored. Nonetheless, there were several notable biochemical differences, including that the 232R PrP had an increased proportion of mono- and un-glycosylated PrP, and mass spec revealed a reduced amount of sialic acid and GalNAc in the GPI anchors of the 232R mice. They inoculated both mouse lines with 7 different human prion isolates. 2 isolates yielded little or no transmission to mice with either wild-type or 232R human PrP; 1 isolate transmitted equivalently to both lines; and 4 isolates transmitted significantly faster to the 232R mice. In some cases, the difference was large — for instance for an sCJD MM1 isolate (called M1 I22 in the paper), the 232R mice died after 300+ days, while the mice with wild-type human PrP took 600 days. In each case, the neuropathology looked similar, it just onset much more rapidly. They concluded that M232R accelerates prion disease in a prion strain-dependent manner.
conclusions so far
While there remain a lot of mysteries, I see enough data here to be fairly confident that differences in the GPI signal sequence, and therefore in the resulting GPI anchor, somehow affect prion disease risk. The human genetics are conclusive that M232R causes increased risk, the question is just by what mechanism. The mouse model work from [Puig 2019] and [Kobayashi 2023] offer the outlines of a mechanism by showing that changing the GPI signal sequence on PrP can result in biochemical changes including a reduction in sialyation. These may in turn lead to other differences such as a reduction in N-linked glycosylation. There may also be a change in subcellular localization, though such changes are usually just anecdotes — it’s hard to gather really statistically robust data in an in vivo system to show how localization is affected. One or more of these changes ultimately affects how fast prions replicate, although it matters more for some strains than for others.
This is all highly plausible. If anything, the problem is there are too many plausible mechanisms, and more than one may be at work, so it’s hard to give one succinct answer as to why the GPI signal matters. For instance, different prion strains seem to “prefer” different N-linked glycosylation states of PrP, so maybe the glycosylation difference is ultimately what matters. But localization is at least as strong a contender: PrP is believed to be localized to cholesterol-rich lipid rafts thanks to its GPI anchor, and given the mechanism of prion replication — PrPSc templates PrPC, differences in local concentration could have a big impact on how often two PrP molecules get to touch each other. If one GPI anchor makes PrP more spread out than another, then prions will replicate more slowly — it’s as if the year were 2020 and they were standing 2 meters apart. And I didn’t even touch on all the hypotheses nominated in these articles, which also touched on things like trafficking into and through the ER. Cell biology of the secretory pathway is complicated, and a lot of the methods for studying it in vivo are low-throughput and imprecise. For now, it seems fairly safe to say that the GPI signal matters, and there are multiple possible reasons why.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.