DNA methylation primer
Dr. Tessa Bertozzi from the Whitehead Institute visited our lab to give us a primer on DNA methylation and epigenetic regulation of gene expression. Here are my notes from the primer.
Epigenetics can be defined as functionally relevant changes to the genome that do not involve changes in DNA sequence. Even though your genome is (to a very close approximation) the same in every cell in your body, the epigenome is very different in different tissues and different cell types. The epigenome influences which genes are “on” and “off” in different cells, what makes a neuron different from a skin cell. Ultimately, which genes are on or off is determined by transcription factors, but the epigenome has a major influence on transcription factor binding.
DNA is not just a loose double helix floating around in the nucleus of your cells. The double helix is wrapped around nucleosomes, which are balls of histone proteins. This complex, called chromatin, is then folded in on itself in a higher-order structure.
There are at least 3 main uses of epigenetic regulation in our cells:
- Suppression of transposable elements [Deniz 2019]
- Genome architecture — 3D organization of chromatin [Dogan & Liu 2018]
- Dynamic regulation and heritable maintenance of transcriptional programs across cellular generations [Waddington 1957]
Two main modes of epigenetic regulation are DNA methylation — the topic of today’s primer — and histone modification, which we do also need to touch on briefly later on.
The main topic of today’s primer is DNA methylation. Unlike histone modifications, which are modifications to proteins that the DNA is wrapped around, DNA methylation means the DNA itself is modified. Specifically, it refers to the addition of a methyl group at the 5’ position of cytosine (C) to yield 5-methylcytosine (5mC), as shown in the diagram below.
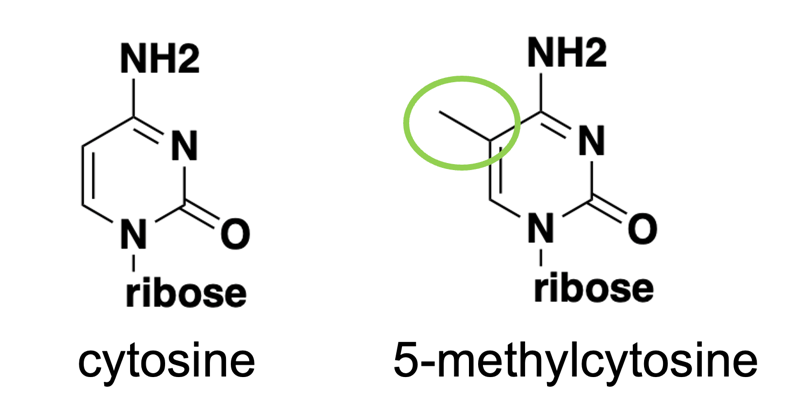
In mammals, methylation of cytosine occurs almost exclusively in the context of a CpG dinucleotide, meaning, a C followed by a G. This is because mammals have 5mC writers that recognize CpG dinucleotides. (There is actually a bit of CpA methylation, especially in the brain, but we won’t focus on this for today). In plants, 5mC writers will write 5mC in all different context.
5mC needs enzymes install it, remove it, and read it [Vukic & Daxinger 2019]. The main writers are DMNT3A and DNMT3B. They need a co-factor DNMT3L, which is catalytically inactive but associates with 3A and 3B to boost their activity. 3A and 3B are expressed most highly during development to lay down initial marks during cell differentiation. But methylation has to be mitotically heritable — every time cells divide, the new strands start unmethylated and need to get methylated. DNMT1 is the enzyme responsible for methylating the daugher (nascent) DNA strand. If DNMT1 is removed, then after a few cell divisions, all methylation is effectively erased by dilution through cell division. There are also enzymes TET1, 2, 3 that actively remove DNA methylation.
Methylation does not change the sequence of the DNA, in the sense that 5mC still pairs with G. Yet it has a profound functional impact by repressing gene expression. The mechanism by which 5mC represses gene expression is still controversial. Some evidence shows that 5mC directly repels transcription factors. Other evidence points to methylation-binding domains (MBDs) in proteins, which only bind methylated DNA, and these proteins in turn repel the transcription factors. Both are true to some degree in some context; the question is the degree to which they are important genome-wide and life-long in vivo [Zhu 2016]. A recent paper provides pretty good evidence that most of the effect is probably mediated by direct inhibition of transcription factor binding, rather than by MBDs [Kaluscha 2022].
CpG dinucleotide is highly mutable, with the C very likely to mutate to a T if the C is methylated and then deaminated, so over millions of years, our genomes have become depleted for CpG dinucleotides. Most CpGs that do exist throughout the human (indeed, mammalian) genome are methylated. Intergenic CpGs are methylated. The methylation of those intergenic CpGs that occur within transposable elements preventing their transcription. CpGs in the middle of genes are methylated, perhaps preventing transcription starting from the wrong place in the middle of the gene. The only place where CpGs are often unmethylated is at the promoters of genes, where there is usually an especially high density of CpGs, known as a CpG island. Indeed, the reason there are more CpGs at promoters is probably precisely because they are unmethylated and therefore less likely to mutate to a T.
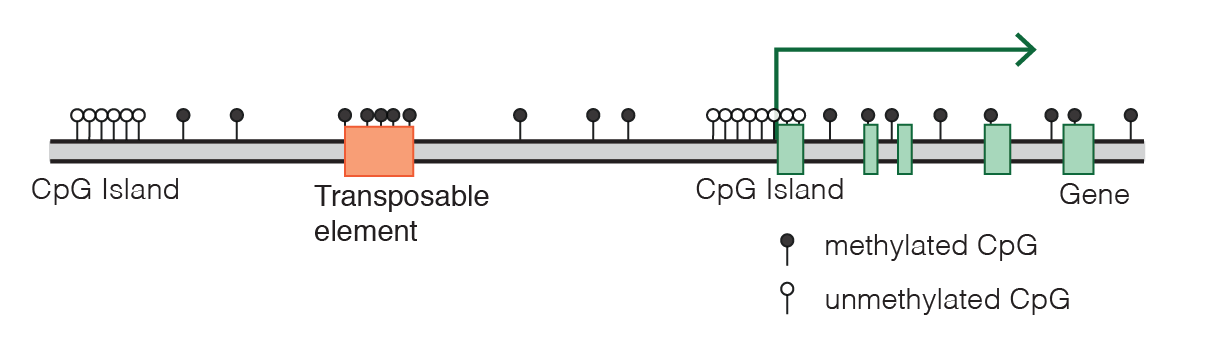
Diagram of CpG distribution in the mammalian genome, by Marius Walter from Wikimedia Commons.
{kind=link}
Only a small subset of CpG island promoters are normally methylated in healthy somatic cells:
- Imprinted genes (where either only the maternal or only the paternal allele is expressed)
- Genes on the inactive copy of the X chromosome
- Germline-specific genes, expressed only in testis or ovary, etc. These are often genes whose continued expression would be detrimental to somatic cells.
In all 3 of the above cases, these are genes we never want to turn back on. Cells generally do not methylate genes that they might want to later reactivate in some developmental context. These genes all get methylated within 4-5 days of development in mouse and 2-3 weeks in human. For genes not falling into the above 3 categories, even if the gene is off in a given cell type or tissue, its CpG island at its promoter is generally unmethylated. Methylation is less easy to turn off and on than histone marks, which polycomb repressive complex (PRC) works on. So histone marks are more often used to turn genes on and off in cell differentiation.
Sometimes, CpG island methylation can also occur as part of a pathological process, for example, silencing of tumor suppressor genes in cancer cells.
CpG methylation changes massively at two points in development. Germ cells initially get almost completely demethylated, then as they differentiate, get highly methylated again, reaching 80% of CpGs in sperm and some lower number in oocytes. Then after fertilization, the pluripotent cells of the embryo again drop to 20% methylation at the blastocyst stage. Then as the embryo differentiates, it acquires a lot of methylation again [Deniz 2019]. The demethylation process in de-differentiated cells is driven mostly by sequestering DMNT1 into the cytosol so that it can’t act on the DNA which is in the nucleus. At least that’s true for the maternal genome; for the paternal genome there is also at least some evidence of direct demethylation by TET enzymes.
We now have a lot of different tools for measuring DNA methylation:
- Methylation specific restriction enzyme-based assays (e.g. HpaII digests). This is very old school and not often used now.
- Affinity enrichment-based approaches (e.g. MeDIP), using antibodies specific to methylated CpGs to pull down DNA and then sequence those pieces.
- Bisulfite sequencing — use of bisulfite to convert C→U (and then U reads like a T in sequencing) at all non-methylated Cs, followed by short-read sequencing. The chemistry of the bisulfite reaction is actually the opposite of the natural mutagenic process in our cells — methylation actually protects C from converting to T by bisulfite, whereas in natural mutagenesis, C is much more likely to convert to T if it is methylated.
- Clonal bisulphite sequencing
- Pyrosequencing
- Whole genome bisulfite sequencing
- RRBS
- Single-molecule nanopore sequencing [Xu & Seki 2020]. Because this allows for long individual reads, you can see how far methylation is spread in cis along a single allele.
While not the main topic of today’s primer, it’s worth briefly touching on histone modifications as another source of epigenetic information. The tails of histones, which protrude from the nucleosome, can undergo several chemical modifications, chiefly acetylation and methylation at different specific amino acids. Some modifications are repressive — they cause the chromatin to become more compacted and less accessible to be transcribed into RNA, while others are activating — they make the chromatin looser and more likely to be transcribed, increasing gene expression. The correspondence of these chemical marks to effects on gene expression has been called the “histone code” and is one major mechanism of gene regulation across different cell types in our body. For example, H3K4me3 (trimethylation of histone 3 residue lysine 4) is an important activating mark at active gene promoters, especially at CpG islands (introduced below). H3K27ac (acetylation of histone 3 residue lysine 27) is another activating mark. H3K9me2/3 and H3K27me3 are important repressive marks. There exists a “HUSH” complex that recognizes self vs. non-self DNA, by determining whether the DNA has introns. Viruses almost entirely lack introns, because they are under selection pressure to minimize genome size. The HUSH complex recognizes this lack of introns and silences the DNA, primarily by H3K9 methylation.
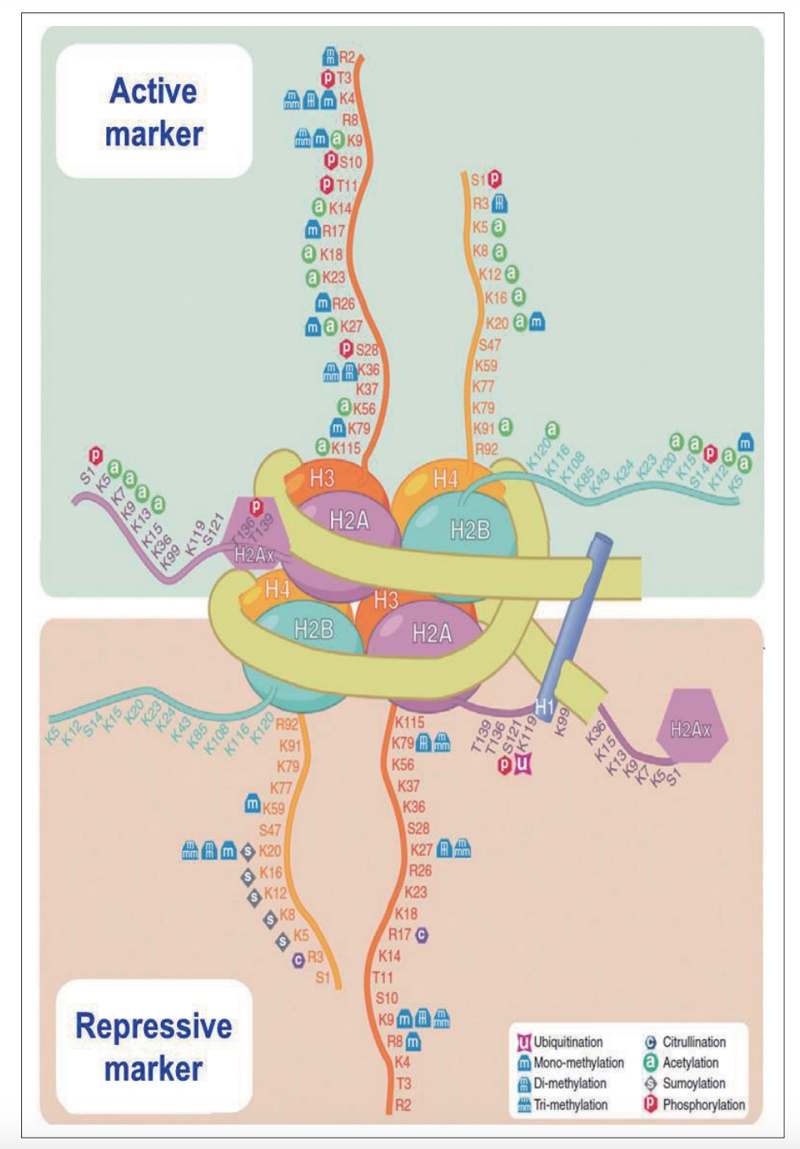
Diagram of histone modifications. Figure 4 from [Kim 2014].
How do histone modifications work? Many of them are marks that need to be read by “readers”, which are other proteins which mediate the functional effects. But histone acetylation, especially H3K27ac, directly physically loosens up chromatin, making the DNA less tightly bound around the nucleosomes, this promoting transcription, apparently without the need for readers to mediate this effect. The reason is that negatively charged DNA binds tightly to positively charged lysines in histones, but acetylation of lysine removes this positive charge, reducing the strength of ionic interaction between DNA and histones.
The main tool for studying histone modifications is still ChIP-seq, chromatin immunoprecipitation with sequencing. Cut&tag and cut&run are also popular alternatives. You use antibodies to pull down DNA bound to histones with certain modifications of interest, then you sequence it.
The de novo methyltransferases 3A and 3B exist in an autoinhibited state at baseline. They need to encounter an unmethylated H3K4 tail (H3K4me0) to become activated. Thus, any active promoter, which usually has H3K4me3, is incapable of activating DMNT3A/B. This mechanism helps protect genes that might need to stay on or re-turn on at some point, from getting DNA methylated. Also, 3A and 3B are only highly expressed in development; in adult somatic cells, they are minimally expressed, though there is often plenty of DNMT1 to maintain methylation. The brain is unusual though, having substantial 3A expression in adulthood.
Our genome is full of endogenous KRAB zinc finger (ZF) proteins [Wolf 2015, Imbeault 2017, Deniz 2019] — there are over 400 in the human genome. There is a lot of variability between species. Guinea pigs have >1000 of them; birds almost completely lack them. The number of ZFPs that an animal has is directly related to its number of transposable elements. They are the result of an evolutionary arms race to silence the transposons. The zinc fingers sequence-specifically bind double-stranded DNA. Transposons evolve to evade the ZF repression, and the ZFs evolve to chase them. In fact, they evolve so rapidly that even two different mouse strains (B6 and CAST) can have profound differences in ZF silencing of transposons [Bertozzi 2020]. The evolutionary pressure comes both from not wanting the transposons to express, but also not wanting them to cause translocations between our chromosomes. ZFs are very repetitive and have only recently been discriminated from each other in the reference gneome. KRAB is a repressive protein domain that prevents transcription. It interacts with a protein known as KAP1 (TRIM28), which is a scaffold protein that recruits myriad other repressors. Among other things, KAP1 recruits HDAC to remove histone acetylation marks, thus compacting chromatin. It also recruits HMTs to cause histone methylation (H3K9me3), which can be read by HP1. Over time, eventually the regions targeted by ZF-KRABs tend to get DNA methylated, but functionally why/how this occurs is still unknown. Our genome also has zinc finger proteins fused to other functional domains, such as domains that activate transcription rather than suppressing it. There are even versions of KRAB domains that are activating rather than repressing. Some ZF-KRABs also include a SCAN domain which contributes to the protein’s function.
The epigenome has been targeted in many therapeutics. There are small molecule inhibitors of DNA methyltransferases and of histone methyltransferases, demethylases, acetyltransferases, and deacetylases, used mostly in cancer. But these are not at all specific to any one gene and generally have poor side effect profiles. There are emerging therapies in preclinical development that target the epigenome in a directed way by using a DNA-binding domain such as a catalytically dead (i.e. unable to make double-strand DNA breaks) Cas9 enzyme from a CRISPR system (dCas9). As of 2023, only one such therapy has gotten to the clinic: the CRISPRa drug CRD-TMH-001 for Duchenne muscular dystrophy [NCT05514249]. That drug used a CRISPR activator (CRISPRa) system consisting of dCas9 fused to VP64, intended to upregulate expression of a full-length isoform of dystrophin. Very sadly, the one patient treated in that trial passed away. It appeared to be due to an immune response to the high dose AAV9 therapy and thus probably does not say much about the viabilty of the CRISPRa approach more generally.
There are additional therapies in this vein — call them epigenetic editors — that are at the technology development or preclinical proof-of-concept stage. In each version, you have some DNA-binding domain engineered to target the promoter of a gene of interest, fused to an effector domain. Most often, the effector domain is KRAB; the DNA-binding domain could be CRISPR guide RNA with a dCas9 (“CRISPRi”, with the “i” standing for interference) [Gilbert 2013], or an engineered ZF, [Zeitler 2019, Wegmann 2021], or a TALE [Mlambo 2018]. It is debatable how permanent any of these might be. Endogenous KRAB-ZFs do cause histone mark changes, but it’s not clear if these are permanent absent continued expression of the KRAB-ZF; the transposons eventually get DNA methylated and are shut off permanently, and expression of the endogenous ZF-KRAB later turns off. But we don’t know how that DNA methylation occurs. DNA methylation has not been shown to occur when KRAB alone is used as a functional domain with an engineered ZF. The engineered ZF-KRAB and CRISPR-KRABs do cause histone mark changes, which don’t persist well in dividing cells; how permanent they might be in post-mitotic cells is not really known. This is due to two technical limitations. First, a lack of ways to only transiently express these tools in neurons — all we have is AAV, which is effectively permanent relative to the lifetime of a mouse. Second, even if we could transiently express these engineered proteins in neurons, a mouse’s lifetime is not long enough to ask whether repressive histone marks installed by KRAB might be lost after 3 or 5 or 10 years absent continued expression of the KRAB. A relatively recent innovation is CRISPRoff [Nunez 2021], which includes domains from the DNMT3A and DNMT3L proteins (without the autoinhibiting parts) to cause DNA methylation, which appears to be more permanent.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.