DNA methylation: durability and what's unique about the brain
Following our DNA methylation primer back in June, Dr. Tessa Bertozzi from the Whitehead Institute returned to our lab to give us a second lecture on DNA methylation, focusing on how the brain is unique compared to other organs. Here are my notes.
DNA methylation has two main roles. One is silencing transposons, which make up half our DNA. The other is regulating genes, though even genes that are “off” in a given cell type or tissue usually have unmethylated promoters; DNA methylation at promoters occurs in cases of imprinted genes, X-inactivation, and germline-specific genes. (DNA methylation can also occur aberrantly, for instance silencing a tumor suppressor in cancer). For a further overview of DNA methylation, see Tessa’s primer or read [Zaidi 2010].
A caveat to most of what’s known and presented here today: because the advent of tools to install DNA methylation is so recent, there aren’t yet a lot of experiments report where someone could perturb the system.
Writing. The writers of DNA methylation are DNMT3A/B, which install de novo methylation, and DNMT1, which maintains DNA methylation. It does so by recognizing hemimethylated DNA, which could arise for instance in the case of cell division and DNA replication, in which the parent strand is methylated and the nascent daughter strand is not. DNMT1 makes the hemimethylated DNA fully methylated. The symmetric nature of CpG — its reverse complement is also CpG, which is not the case for C in any other dinucleotide context — is important to the mechanism by which it is inherited on cell division.
Erasing. DNA methylation can be lost on cell division, simply by dilution, if DNMT1 is not there to maintain and reinforce it. This is what happens in embryonic development, when DNMT1 is sequestered to cytosol and DNA methylation is lost. There are also erasers of DNA methylation via active removal of methyl groups: the TET enzymes (this is a bit of an oversimplification, see below). TET stands for ten-eleven translocation, named after a chromosome 10/11 translocation denoted t(10;11)(q22;q23) that causes acute myeloid leukemia by fusing TET1 to MLL [Lorsbach 2003]. We have TET1, TET2, and TET3.
Reading. Mammals have 5 different DNA methylation readers: MeCP2, and MBD1/2/3/4 [Van den Veyver & Zoghbi 2000, Bogdanovic & Veenstra 2009]. MeCP2 is important in the brain.
Primordial germ cells (PGCs) are very difficult to transduce with AAV, and they undergo almost complete demethylation (after which sperm and egg get differentially remethylated) [Deniz 2019, Greenberg & Bourc’his 2019]. This means that from first principles there’s very little risk of germline DNA methylation with CHARM therapeutics.
As a refresher, recall that CHARM uses a DNA-binding domain, D3L, and an unmethylated histone tail H3K4me0 to recruit and activate endogenous DNMT3A [Neumann & Bertozzi 2024]. CpGs overlapped by the CRISPR guide or zinc finger can be missed because the DNA-binding domain sterically occludes the methylation from being installed, but you nevertheless get nearly complete silencing. AAV DNA sticks around as an episome in transduced cells, apparently for at least several years, possibly forever, based on human & animal data [Marco 2021, Muhuri 2022]. CHARM can be designed to self-silence, which could theoretically have advantages in terms of reducing off-targets and immunogenicity concerns. If we design CHARM to self-silence, will the DNA methylation it installed in neurons remain durable indefinitely? Or is there a risk that the methylation will be lost and we’d be better off designing our CHARM to constitutively express as long as the AAV is present?
Data from Human Protein Atlas show that both writers and erasers of DNA methylation are expressed (at the protein level) in the brain and specifically in neurons:
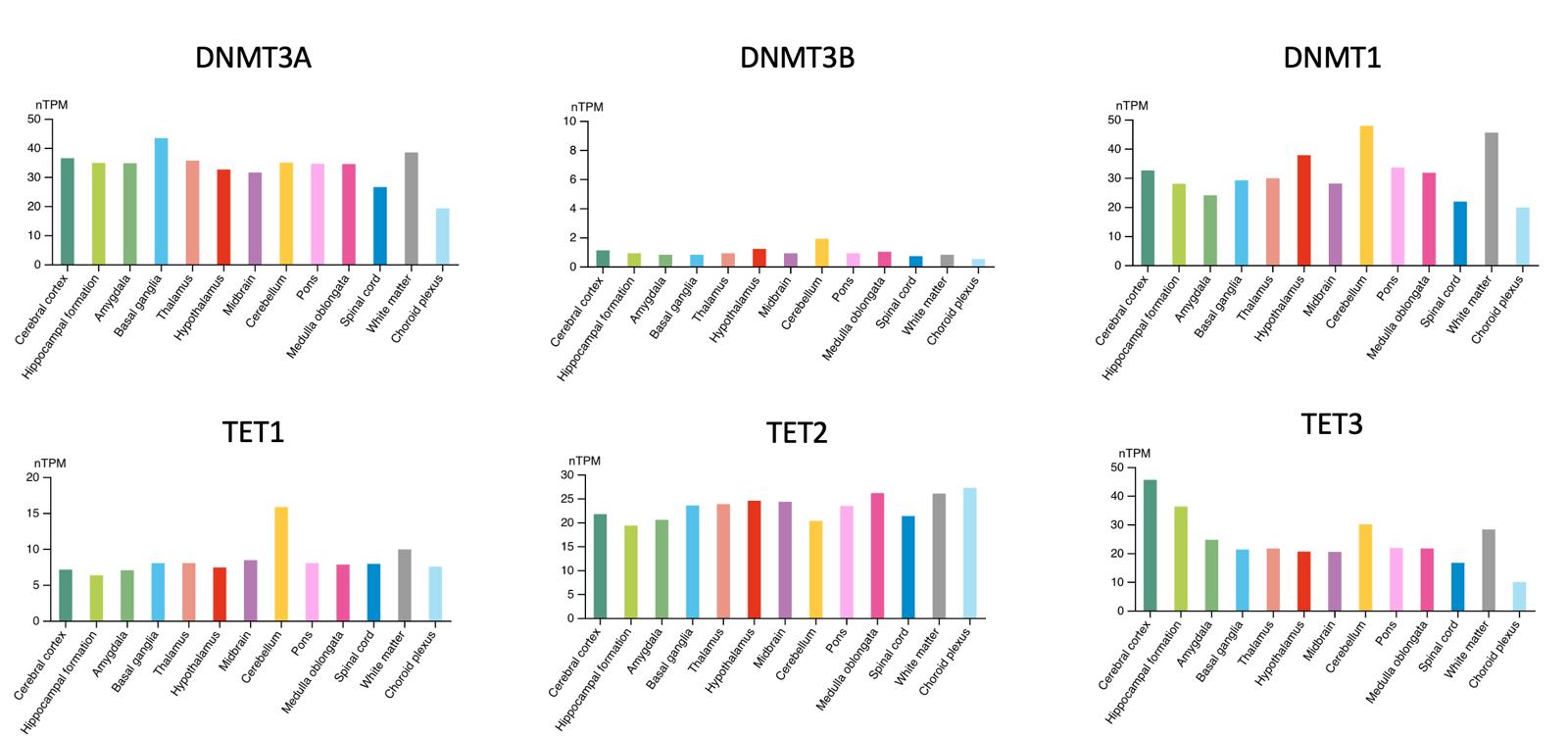
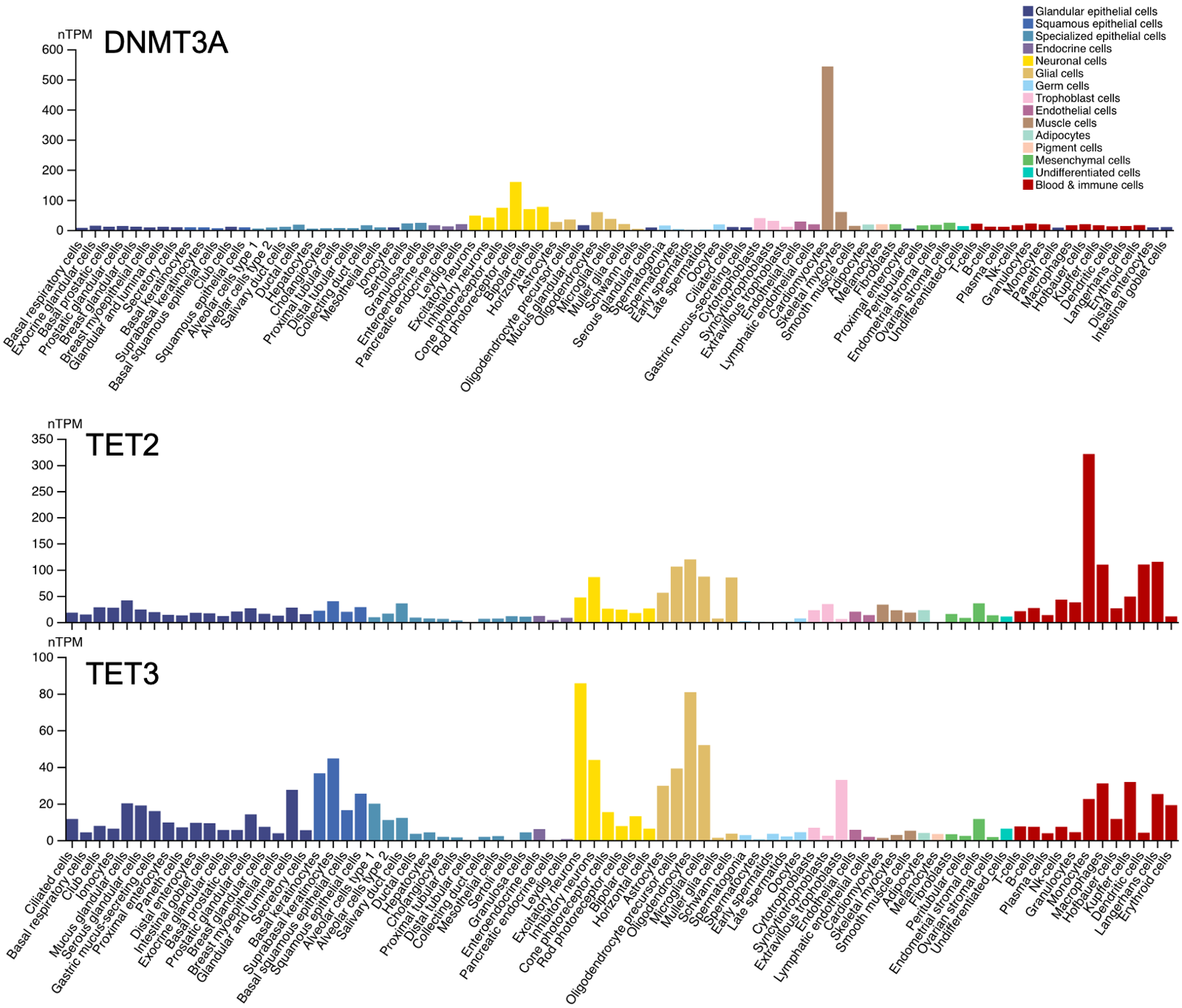
A caveat is that the y axes are not identical in all the above plots (look carefully and you’ll see). But the overall impression the plots give is not wrong: in the brain, DNMT3A is expressed while DNMT3B is minimal; all of the TET enzymes are expressed at some reasonable level.
All of these enzymes are important and cause severe disease if lost. There are human Mendelian diseases caused by loss of DNMT1 [Klein 2011, Winkelmann 2012]. A mouse double conditional knockout of DNMT3A and DNMT1 found resulting cognitive deficits [Feng 2010]. Critically, this was associated with a long-term decrease in neuronal DNA methylation. Although back then, the 5mC content could only be assessed by mass spec and by targeted bisulfite sequencing; whole-genome bisulfite sequencing was too new and was not yet used to systematically examine where in the genome the methylation was lost. There are a lot of mouse studies showing that knockout of TET1, 2, or 3 causes defects [reviewed in Antunes 2019]. Finally, MeCP2 loss-of-function mutations cause Rett syndrome [Van den Veyver & Zoghbi 2000], apparently acting through as many as 4,189 genes that it regulates [Liu 2024]. Rett syndrome only affects girls, because MeCP2 loss-of-function is fatal on an XY karyotype background, while in XX it affects just half their cells due to random X-inactivation.
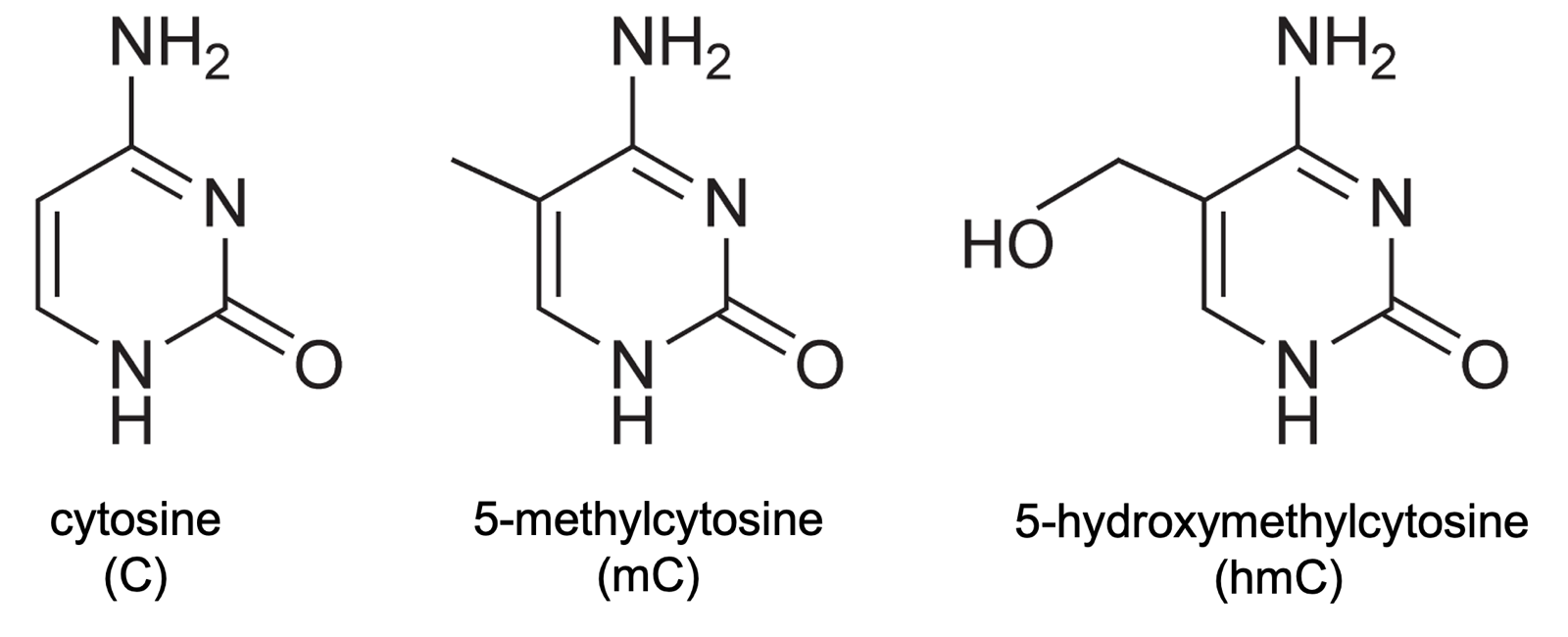
Besides mC, cytosine can also become 5-hydroxymethylcytosine (5-hmC or simply hmC), pictured above. In addition, while the percentage of CpGs that are methylated, genome-wide, is far higher, C can also be methylated in other dinucleotide contexts. Both of these phenomena occur especially in the brain [Jang 2017].
In the shorthand of DNA, “H” means A, C, or T, so non-CpG methylation is abbreviated mCH. Here are some summary data on how common these modifications are, from [Clemens & Gabel 2020]:
| dinucleotide context | non-neuronal | neurons |
|---|---|---|
| CpG | 80% mC, 3% hmC | 40-65% mC, 15-40% hmC |
| CpH | 0.02% mC, 0.02% hMC | 1.5-3% mC, 0.02-1% hmC |
As you can see, neurons are unique in having less mCpG and much more hmCpG, and more modifications to CpH overall. Most of the CpH is CpA. CpH are much more common than CpG, genome-wide, therefore despite the smaller percentages, mCpH is actually just as prevalent in neurons as mCpG.
While mCpG is established very early in development and maintained throughout, mCpH accumulates gradually over development, plateauing by 16 years old in humans or 4-6 weeks in mice [Lister & Mukamel 2013]. mCpH acquisition appears to be associated with synaptic density and DNMT3A expression.
Earlier we simplified by saying that TET enzymes actively remove methylation from cytosines. In fact, it’s more complicated. TET only catalyzes oxidation reactions from mC to hmC, which can then continue on to 5-formylcystosine (5fC), 5-carboxylcytosine (5caC) and other species that get recognized for base excision repair (BER) and replaced with an unmodified cytosine. Therefore, if TETs are in the act of removing methylation marks, you might expect to see more hmC.
Above we saw that TETs are expressed in the brain and in neurons in particular, and neurons have elevated hmC. At a glance, you might worry this means there is random erosion of 5mC marks in neurons over time. But in fact, it’s not random: the genomic positions where hmC and mCpH marks appear in neurons is highly conserved and reproducible across individuals [Szulwach 2011]. It is thought that it mostly affects genes involved in learning and memory. So it may be that a particular gene, say PRNP, that you target and methylate, would have no reason to be subject to erosion of methylation over time.
So, the million dollar question: will CHARM prove durable over the human lifetime?
The reasons for pessimism:
- Neurons are post-mitotic, which means there is no cell division to maintain and reinforce DNA methylation.
- Neurons express TET, so active demethylation could accrue over time, and indeed, high levels of 5-hmC in neurons could reflect the fact that this occurs. Indeed, dynamic changes in DNA methylation in neurons have been reported.
The reasons for optimism:
- Neurons are post-mitotic, which means there is no risk of methylation loss through either replication errors or dilution, both of which would otherwise be risks [Dmitrijeva 2018, Ming & Zhang 2020]
- Erosion of mC to hmC is highly PRNP is a specific process. Arguably, PRNP is unlikely to be targeted for demethylation, and is not near any highly transcribed genes.
- Some demethylation could be tolerated before PRNP reactivation would occur.
- A properly tuned self-silencing CHARM construct could be designed to reactivate before PRNP reactivates. The promoter driving CHARM would need to be designed to be slightly more prone to reactivation than the PRNP promoter.
Our level of optimism vs. pessimism matters, because it feeds into a decision of whether, and how aggressively, to self-silence CHARM. If you think methylation may not be permanent, you want to constitutively express CHARM. If you are confident it is permanent, then you might self-silence it if you have reason to believe this would help manage off-target or immunogenicity risks.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.