AAV primer

Ken Chan from the Deverman Lab at the Broad Institute visited our lab to give us a primer on AAV. Here are my notes.
Today we have a really impressive arsenal of gene-based medicines — gene replacement, base editing, prime editing, tRNAs, CRISPRoff, CHARM, and so on — that could theoretically treat or cure almost all genetic diseases if you could get them into the right cells. Delivery — the task of getting your therapeutic gene of interest into those cells — is the principal challenge for genomic medicine. Delivery requires packaging DNA or RNA in a shell that protects it from degradation by nucleases, evades immune response, attaches to the surface of the correct cells, get the genetic material internalized into the cell, and then get it localized into the correct compartment (cytosol for RNA delivery, or nucleus for DNA delivery). Viruses evolved to do all of these things. Therefore, viruses are the obvious place to look for a way to deliver genes therapeutically.
AAV was discovered nearly 60 years ago in preparations of adenovirus [Atchison 1965, Hoggan 1966]. Researchers trying to isolate adenovirus found small DNA virus particles that could only replicate in the presence of adenovirus. They called them adeno-associated viruses (AAV). The adenovirus came to be referred to as a helper virus. Despite the name, though, AAVs can piggyback off of multiple different viruses in order to replicate: adenovirus (Adv5) but also herpesvirus (HSV-1, HSV-2, HCMV, HHV-6, V2V), papillomaviruses (HPV-16), and bocaviruses (HBoV1). The helper viruses serve to inhibit antiviral pathways, and get the host cell into a state favoring viral replication. The serotypes such as AAV2, AAV6, etc. are just distinct groups of antigen presented on the surface of the capsid, a functional distinction based on antisera reactivity. But serotypes are not actually perfectly correlated with phylogeny [Gao 2004]. There are about 13 serotypes, while phylogenetically there are dozens of types grouped into “clades”, and serotype does not correlate well with clade; sometimes distinct serotypes (e.g. AAV1 and 6) are actualy very closely related.
Several features of AAV that make it an attractive delivery vehicle for delivering genes into cells:
- Non-pathogenic
- Replication incompetent
- Transduces both dividing and nondividing cells, in vitro and in vivo.*
- Simple genome - only need to retain the ITRs, can package anything in between
- Genome will stabilize and not degrade, so you get long-term episomal expression without host genome integration
- Can be engineered
- Many naturally occurring serotypes to start from
- Non-enveloped.
*Getting into dividing cells is easier because the nuclear membrane breaks down during cell division. AAV has the ability to get DNA into the intact nucleus of a non-dividing cell.
By contrast, for example, lentivirus has integration risk, and does not stay episomal.
AAV is non-enveloped, it’s just a protein shell which makes it clean to purify and manufacture. In contrast, enveloped viruses get their envelope from host cell membrane at the moment of egress from the cell, and they take with them whatever proteins are stuck into that membrane. This makes it much harder to control their composition in manufacturing.
People have been trying to use AAVs for gene therapy since the early 1990s for cystic fibrosis. They shot AAV encoding CFTR into the lungs of rabbits and were able to detect expression [Flotte 1993], which led them only 2 years later to launch a Phase I trial in 25 patients [Flotte 1996]. The trial results looked fairly safe but also provided no evidence of disease modification [Flotte 2003]. We now think that this was probably due to all of the following factors:
- a non-optimal serotype — they used AAV2, which doesn’t transduce lung cells all that efficiently
- too low a dose (and inability to re-dose due to immune response)
- physical barrier of air — the lung was not the easiest tissue in which to start because you had to get a liquid preparation of AAV to travel through gas to disperse to the targeted cells
While we know a lot more now, there are still enormous challenges with AAV.
Neutralizing antibodies can affect the efficiency of AAV transduction, and high doses of AAV elicit immune response leading to complement activation that can affect transduction. This is a major challenge in administering AAV.
The gene you’ve delivered could have immunogenic properties. The DNA can form structures that elicit immune response. Cas9, a bacterial protein, may be immunogenic. A gene that the patient was lacking — in the case of gene replacement for a loss-of-function disease — may be immunogenic for that patient even if it’s a human gene that other people have. These things can happen even with intracellular proteins, due to toll like receptors and so on.
Both of the above are hard to model in any non-human species.
There are also factors that can affect AAV durability:
- Cell division can dilute the genome. This is especially true because if you have multiple AAV genomes in the same cell, they can concatemerize, such that even if you started with >1 copies, still only 1 daughter cell will receive any copies.
- Epigenetic modifications can occur on the episomal DNA, silencing it [Loser 1998, Das 2022, Gonzalez-Sandoval 2023].
Despite all these challenges, there have now been successes. The first FDA-approved AAV gene therapy was voretigene neparvovec for RPE65 blindness in 2017 [FDA press release]. Voretigene neparvovec was the low hanging fruit in terms of all of AAV’s challenges: the amount of AAV needed for retina was very small, the serotype used (AAV2) transduces retinal cells pretty well, and the retina is immune privileged.
Today there are 6 FDA- or EMA-approved AAV therapies, all using naturally occurring AAV serotypes:
| drug | gene | indication | AAV serotype | route of administration |
|---|---|---|---|---|
| voretigene neparvovec | RPE65 | Leber congenital amaurosis | AAV2 | retina |
| onasemnogene abeparvovec | SMN1 | spinal muscular atrophy | AAV9 | systemic* |
| eladocagene exuparvovec | AADC | AADC deficiency | AAV2 | intraparenchymal brain injection |
| etranacogene dezaparvovec | F9 | hemophilia B | AAV5 | systemic |
| valoctocogene roxaparvovec | F8 | hemophilia A | AAV5 | systemic |
| delandistrogene moxeparvovec | DMD** | Duchenne | rh74 | systemic |
*intrathecal has also been evaluted in trials but is not yet approved
**microdystrophin
Last year, a press release announced that >3,000 children had receieved onasemnogene abeparvovec, and that there was 100% response rate out to 7.5 years post-dose among children who received it before symptom onset. This provides some indication of long-term durability of AAV.
Given the compelling efficacy of some of the above therapies, why can’t we do the same thing for all other genetic diseases? The biggest bottleneck is efficient targeting of the right organs and right cell types. Naturally occurring serotypes just have too low of efficiency. Effective therapies for a great many diseases remain to be unlocked by engineered AAV vectors that can efficiently transduce the right cells in older children or adults.
The atomic structure of AAV [Xie 2002] was hugely enabling for engineering efforts. The sequences that the Deverman lab modifies in their engineered vectors are located in exposed loops on the capsid surface.
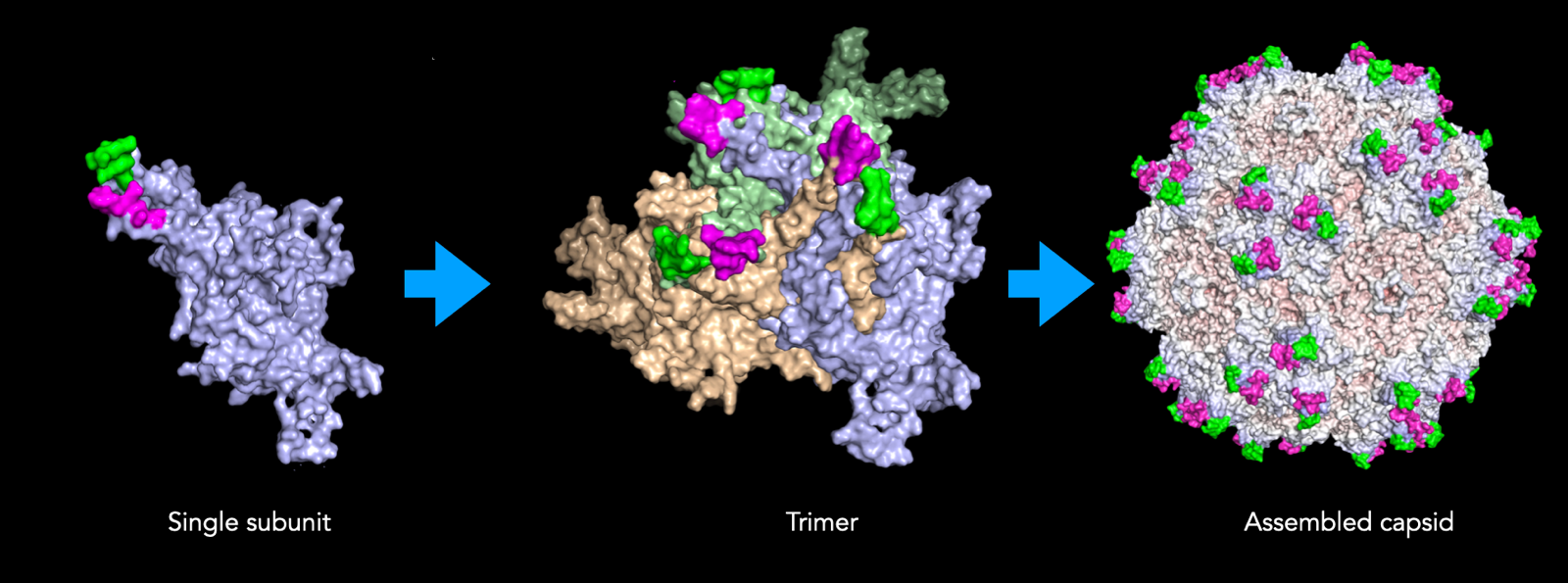
Ben Deverman made an early decision to focus on these exposed loops and insert novel peptides there to pan for capsids with novel targeting properties, resulting in the discovery of PHP.B [Deverman 2016].
A complete AAV capsid is made up of 60 individual subunits. The way these 60 subunits are assembled yields geometrical features with axes of 2, 3, or 5-fold symmetry:
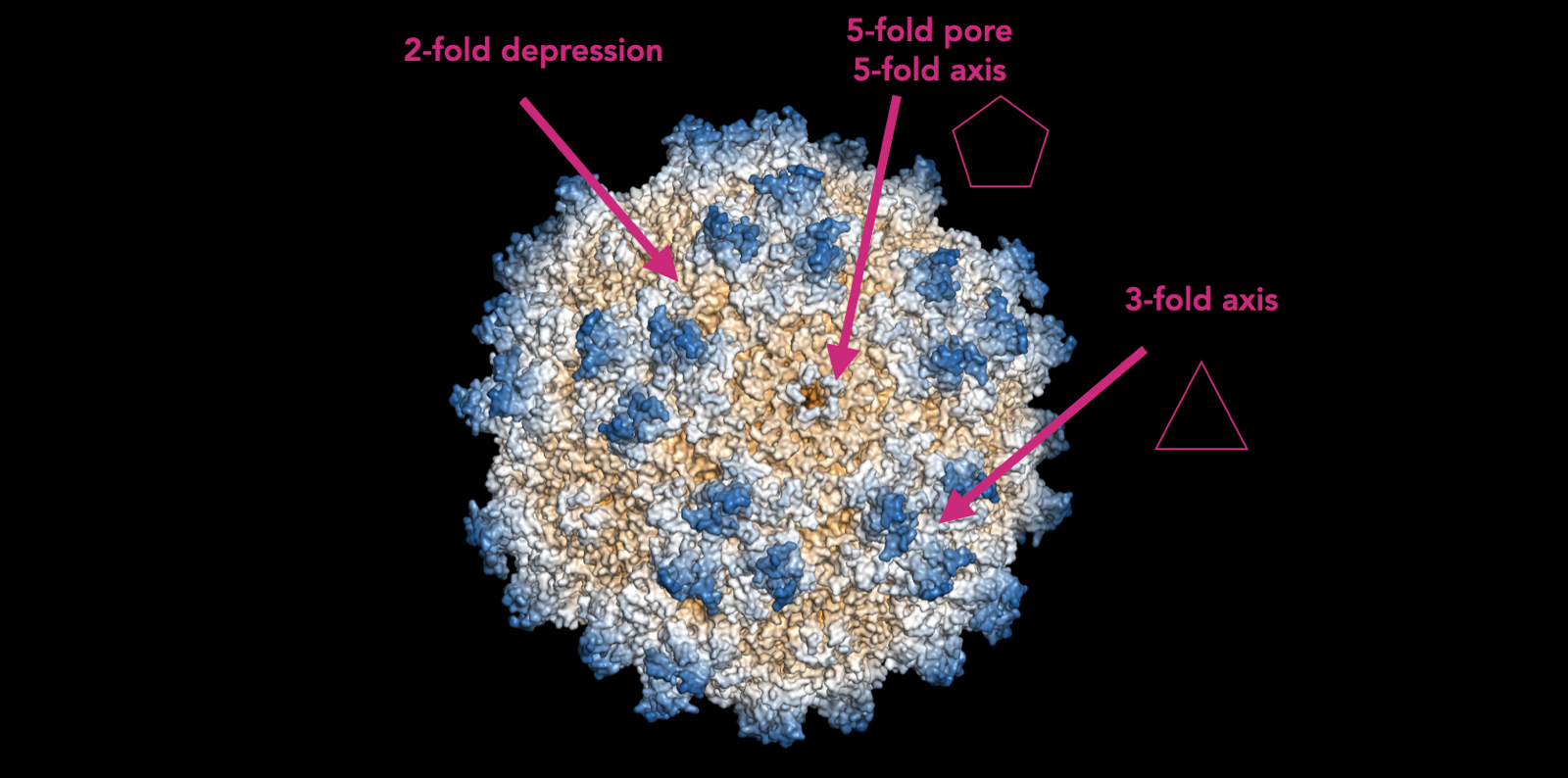
Each of these is functionally important. The 3-fold axis is where the loops that are modified in engineered AAVs are located. The 2-fold depression is where most antibodies bind, determining AAV serotype. The 5-fold pore is where the DNA being packaged into it gets inserted. There is a protein that attaches to the pore and ratchets the double helix through the pore. So the whole capsid is assembled first and then the DNA is inserted. There are many 5-fold pores on the surface of one capsid, and no one knows how the AAV capsid “knows” not to push more DNA into one pore once DNA has gone in through a different pore. Thus, at present, we can only manufacture AAV in cells, as a biologic. If you someone could figure out how to insert the DNA at 1 genome per capsid after the capsid is formed, that would be hugely enabling and might make it possible to manufacture purely synthetic AAV.
The full sequence of the AAV genome was determined in the early 1980s [Samulski 1982, Srivasta 1983], and the ability to make recombinant AAVs with a gene of interest inserted followed soon after [Hermonat & Muzyczka 1984]. Even though AAV had been known since the 1960s, it would have been impossible to use AAVs for gene therapy without knowledge of the genome.
The wild-type AAV genome is simple and elegant:
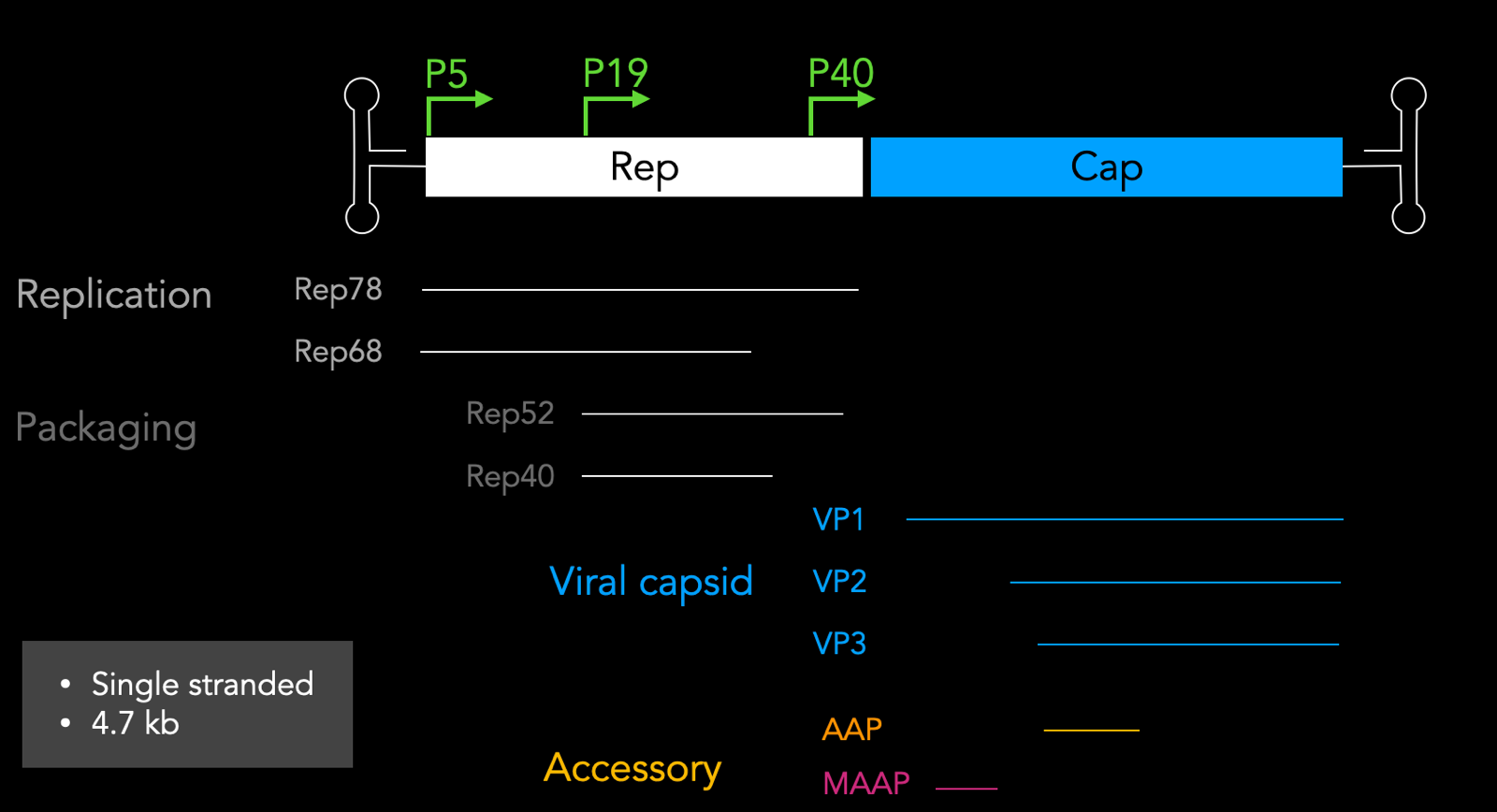
This diagram is to scale and VP1, VP2, and VP3 are all in the same reading frame. They are different splice isoforms with different start codons, all in the same frame. Of the 60 subunits, there are 5 VP1, 5 VP2, and 50 VP3. You can form a capsid with just VP3, so why do you need VP1 and VP2? VP2’s function is still not fully understood, but VP1 has a key pH dependent conformational change that allows the capsid to escape the endosome.
The Cap portion of the genome also encodes 2 other proteins in a different reading frame: AAP and MAAP. AAP is assembly-activating protein which facilitates capsid assembly, and MAAP which was discovered very recently [Ogden 2019]; it associates with the membrane and may be involved in escape but its function is not totally clear.
The Rep portion of the genome has a self-regulating property. The P5 transcription initiation site turns on Rep78 and Rep68, which then go and activate P19, leading to transcription of Rep52 and Rep40.
Inverted terminal repeats serve at least 4 purposes:
- Origin of replication
- Encapsulation
- Gene stability and episome formation - because they loop back on themselves they protect against exonucleases
- Viral genome integration - AAV can actually genome integrate into a few very specific well-defined sites in the genome, but only if Rep is present
To place your own gene of interest into an AAV, simply clone it in between the ITRs, and express the Rep and Cap in trans, in a separate plasmid lacking ITRs. You also need a helper plasmid with E2A, E4A, and VA, which are the essential helper components that wild-type AAVs get from the other viruses they depend on; they inhibit antiviral pathways and stabilize the genome. Thus you end up with triple plasmid transfection: gene of interest, rep cap, and helper.
The structure of a gene of interest (GOI) to package in an AAV can look like this:
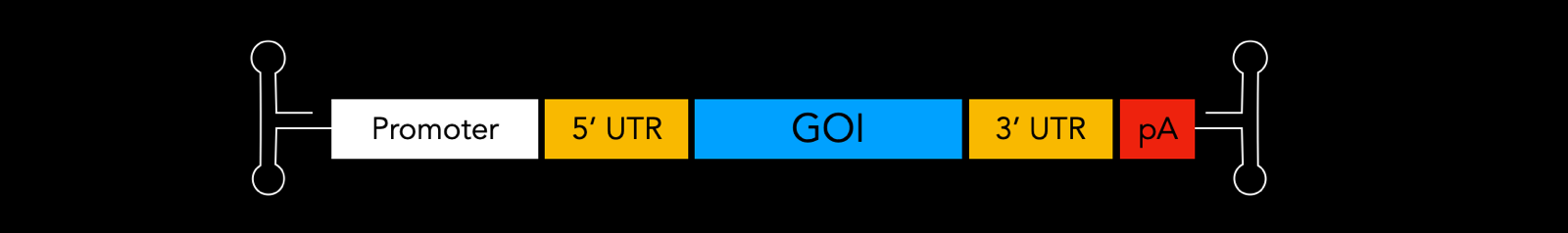
People use many different promoters in GOIs. These can include inducible ones like TET, cell type-specific ones to restrict expression to desired cells like hSyn, GFAP, MECP2, or broad spectrum promoters like EFS, EF1a, CMV, Ubc. Inclusion of introns in the GOI can help to stabilize the mRNA and increase efficiency of mRNA export from the nucleus. In the UTRs, uORFs and miR target sites can be used to regulate or restrict expression. The polyadenylation signal (polyA or pA) helps with mRNA stability, nuclear export, and translation efficiency.
In sum, AAVs are a 25 nm non-enveloped viral capsid that can be used as a versatile single-stranded DNA (ssDNA) vector with a capacity of 4.7 kb.
Q&A
Q. What factors tell the host cell that AAV DNA is foreign?
A. Initially it is single-stranded and non-methylated, both of which are unusual. But once AAV ssDNA has entered, it actually hijacks cellular machinery to make it double-stranded and chromatinized. Indeed, AAV has + and - strands which are both present in manufacturing, and we think that half of capsids get filled with + and half with -. In cells that are doubly transduced, by one of each, they will actually become double stranded. However, people do also sometimes manipulate AAV structure to try to maximize its looped-in-on-itself-ness, because they think that conversion to dsDNA is a rate-limiting step. AAV transduction also requires a Golgi apparatus receptor called AAVR, which helps the DNA out from the Golgi and into the nucleus. AAVR knockout is very difficult to transduce. The rate of uncoating can matter in host cell recognition.
Q. What limits the size of cargo to 4.7kb?
A. Literally the fit of all the DNA in there.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.