Accelerating gene therapy clinical trials in seropositive patients

Parent Project Muscular Dystrophy (PPMD) organized a 1-day symposium at the Broad Institute entitled “Accelerating gene therapy clinical trials in seropositive patients,” focusing on AAV therapy in AAV seropositive patients. These are my notes. These talks were very fast and my notes likely contain errors — if you spot one, please contact me. The video of the event is available here.
Welcome Remarks | Michelle Rengarajan, Harvard Medical School & Mass General Brigham
Dr. Rengarajan (pictured at top) is an endocrinologist at MGH, part of the Ladders to Cures initiative at Broad, and a parent of two Duchenne boys. She is wearing all of these hats today. The reason for today’s symposium: AAV is the mainstay of gene therapy, but half or more of potential patients are seropositive, and we currently cannot re-dose AAV therapies.
Clinical trial experience in patients with pre-existing antibodies to AAV | Kathy High, Rockefeller University
Dr. High’s talk will present the experience up to now with administering AAV to seropositive therapy in clinical trials, and what happens later on with patients who do not qualify for the trials.
Some pre-existing antibodies occur as a result of wild-type virus infection, and are generally lower titer than the neutralizing antibody (NAb) titers that arise after treatment with a vector. Naturally occurring titers and re-administration of a therapy are 2 different problems for therapeutics.
There are a wide range of reported numbers as to what percent of people have pre-existing natural immunity. This is both because the populations in these studies differ, and because the assays differ. For example the below data from [Kruzik 2019] shows 5 studies with a range of 32-63% seropositive. But most have low titers (≥1:40) and many are not cross-reactive against other serotypes:
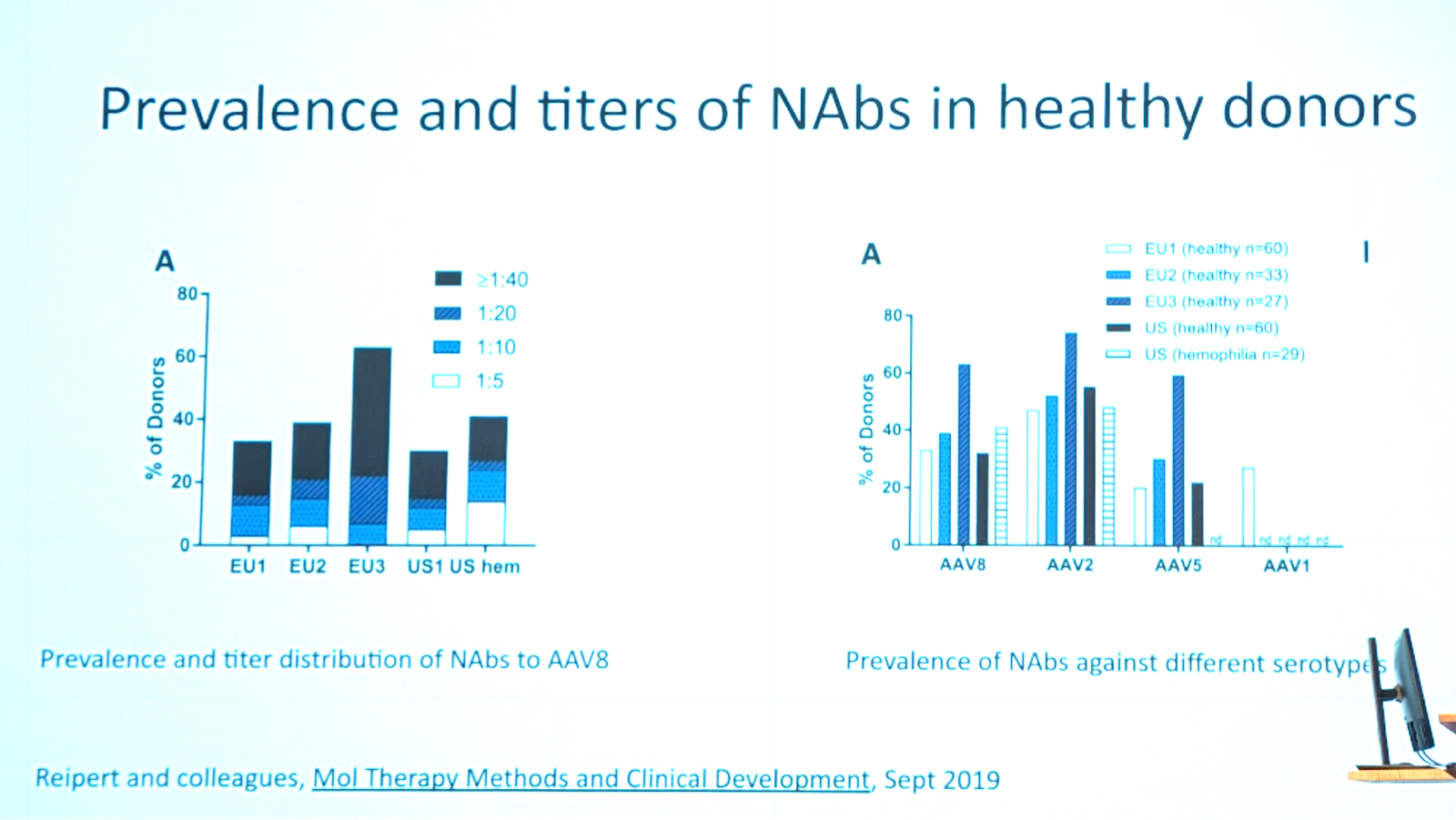
In contrast, in patients who received an AAV2 gene therapy, 100% had very high titers even 14 years post-dose [George 2020].
For direct injection, even high NAb titers do not prevent transduction [Kay 2020]. This was also true in the retinal injections for voretigene.
People had tried using human IgG administered to mice to try to simulate pre-existing NAb titers in a mouse model. They could show that it blocked transduction for intravenous treatment. However, for the first intravenous AAV trials, we didn’t know quantitatively where to set the titer cutoff. Therefore we did admit patients with nonzero titers into a Factor IX liver trial. They compared two titers; the patient with the low titer (1:2) had detectable Factor IX for weeks after, while the patient with the high titer (1:17) never got therapeutic levels of Factor IX exposure [Manno 2006]. Come to find out, there were two similar therapies in development, but one had 100% full capsid while the other had a high rate of empty capsids. They did a mouse study and figured out that, controlling for the same dose of full capsid, injecting a 10X excess of empty capsid actually dramatically helped transduction [Mingozzi 2013]. It appears that the empty capsid acts as a decoy, sopping up the NAb so that the full capsids can transduce. There were similar findings in NHP with natural pre-existing NAb titers. But the problem was just transduction, or in other words, efficacy — there was no evidence of a safety concern when the AAV recipient is seropositive. The FDA summary basis for action for etranacogene breaks down Factor IX activity by pre-existing NAb titer. It is 42% in the low titer patients and 27% in the medium titer patients, both of which are therapeutic levels. Only in the 1 very high titer patient, the Factor IX restoration was just 1.5% which is not enough.
Potential solutions to pre-existing antibodies are:
- Change the product
- Increase particle dose (expensive, but empties may be cheaper)
- Engineer the vector to evade NAbs
- Change the recipient
- Plasmapheresis [Montelheit 2011]
- IgG-cleaving endopeptidase [Leborgne 2020]
- Ablate B cells and plasma cells. A three-drug regimen of rituximab, cyclophosphamide and prednisone trialed at MGH was reported to completely abolish antibody titers in 100% of patients [Zonozi 2021]. That study (which was in patients with an autoimmune disease, not AAV-related) went on 24 months, but for AAV you would only need a much shorter regimen.
AAV Gene Therapy: Caregiver Perspective | Annie Ganot Solid Biosciences
Solid was founded by the Ganot family in 2013 in response to their son’s Duchenne diagnosis. They initiated an AAV trial in 2019, only to find that their own son was excluded due to seropositivity. They are now in Phase I/II (NCT06138639) with a second-generation AAV-vectored microdystrophin construct called SGT-003. They are also committed to solving the seropositivity problem.
Q&A
Q. Given the options Kathy High talked about, such as plasmapheresis and B cell ablation, why can’t we already use those tools to dose patients like your son?
A. We and many other companies are trying to solve this problem, and we feel we are not there yet but it may be in the near future.
Q. Do you see any patient education gaps we need to address?
A. We’re lucky that the Duchenne community is incredibly activated and networked. People generally have high awareness of the issues. A special challenge is for families with multiple affected sons, how to stage their participation in trials. Some families will have their older son participate in a trial today, while they hold out for the older, less-affected son to remain seronegative and receive a next-generation, better AAV therapy in the future.
Overview of human immune responses to AAV | Federico Mingozzi, Nava Therapeutics
Here is Dr. Mingozzi’s timeline of immunity issues in AAV therapy:
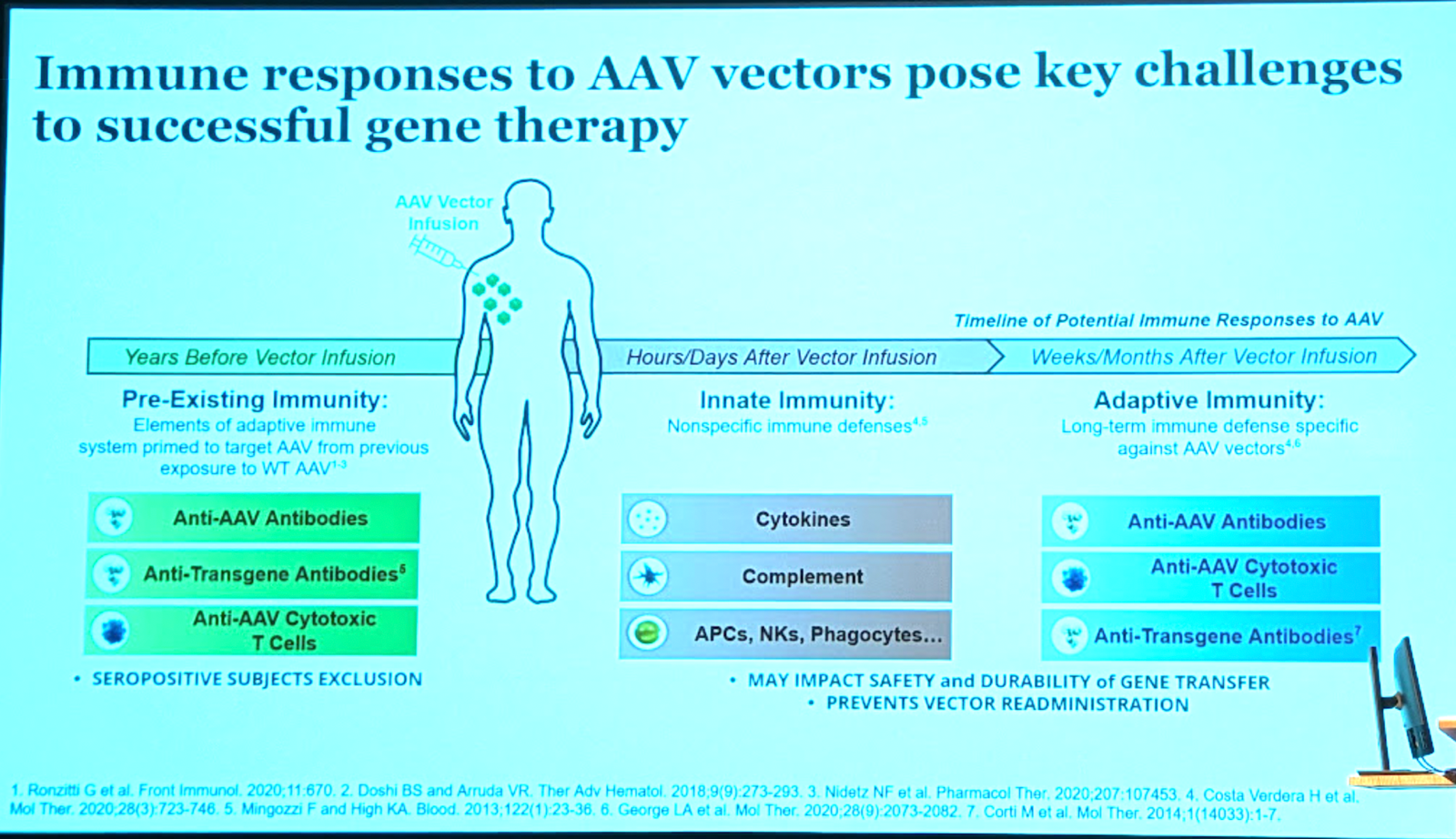
The impact of pre-existing immunity depends a lot on route of administration (there are many - see [Zhu 2021]) and vector dose (higher doses can overcome some titer of NAbs [Mingozzi 2013]). This is analogous to some biologics where you just have to dose higher to overcome anti-drug antibodies.
Here is a diagram of the mechanisms of anti-AAV immunity — both TLR [Monahan 2021] and complement [Wright 2020, Smith 2022] play a role:

Complement is generally involved in pathogen recognition and we would expect it plays a role in AAV immunity. Some studies but not others observe complement activation [Smith 2022]. Both empty and full capsid trigger complement activation. Many drugs are available to counteract complement activation — C5 and C1 inhibitors.
Here is what we know about the mechanism of T cell response to AAV [Mingozzi & High 2011]:

Some general principles of immunomodulation in AAV gene therapy. The first agents to be used were corticosteroids. Some T and B cell targeting drugs have also been used. Ideally you want a short immunosuppressive regimen and it should be targeted, meaning pathway-specific. The field has struggled to find a generalizable immunosuppressive regimen, in part because AAV is used to treat many different diseases, some of which have inflammation as part of the phenotype in the first place. There are also vector-specific considerations. There might never be a one-size-fits-all.
In a UGT1A1 gene therapy trial (a bilirubin disorder, see below), they used sirolimus and prednisone to control immune response, and it was pretty successful [D’Antiga 2023].
Q&A
Q. Can you package an AAV into an LNP to protect if from NAbs?
A. It’s an interesting idea. We explored adding a bilayer in the form of extracellular vesicles, and we do see some protection from neutralization. The process is not very efficient though. Packaging nucleic acid into LNP takes advantage of the negative charge on the nucleic acid; for complex cargoes like AAV it’s not easy to package.
Q. What about rapamycin?
A. Rapamycin is the same thing as sirolumis. It’s been used in combination with prednisone, for several months post-AAV infusion. It’s also been used in combination with rituximab.
Strategies to Modulate AAV Specific Adaptive Responses | Shari Gordon, AskBio
Dr. Gordon is senior director of immunology at AskBio.
We want to make the naturally seropositive patients eligible for AAV therapies. But one concern is even if you can find a way to dose patients who have pre-existing immunity, will they have an even larger secondary response to the AAV therapy than seronegative patients already do? What is the risk both to safety and efficacy?
They dosed healthy volunteers with empty AAV8 capsids. They only enrolled subjects screened as seronegative for AAV8 neutralization. Not surprisingly, all patients developed high titer binding Abs and high titer NAbs. The T cell response was also very strong within 1 week of dosing in 5/8 individuals based in interferon gamma elispot. The response had increased breadth and targeted multiple epitopes and was primarily CD4+ T cell driven. When they looked back they found that the patients with the highest T cell responses had pre-existing NAbs against other AAV serotypes, such as AAV2, even though they had initially been seronegative for AAV8. They think an immunosuppressive regimen to manage both T and B cell response will be needed. They are testing strategies in transgenic mice with AAVrh32.33 and in NHPs.
Immunoglobulin cleavage enzymes cut antibodies into Fa, Fb, and Fc fragments, and are clinically validated - see for instance imlifidase (NCT06241950, NCT06518005). One downside is they are bacterial enzymes and may themselves cause immunity. Another option is FcRn inhibitors, which bind the neonatal Fc receptor (FcRn; FCGRT) and prevent antibody recycling so the Ab is degraded. They studied these two approaches head-to-head in an animal model.
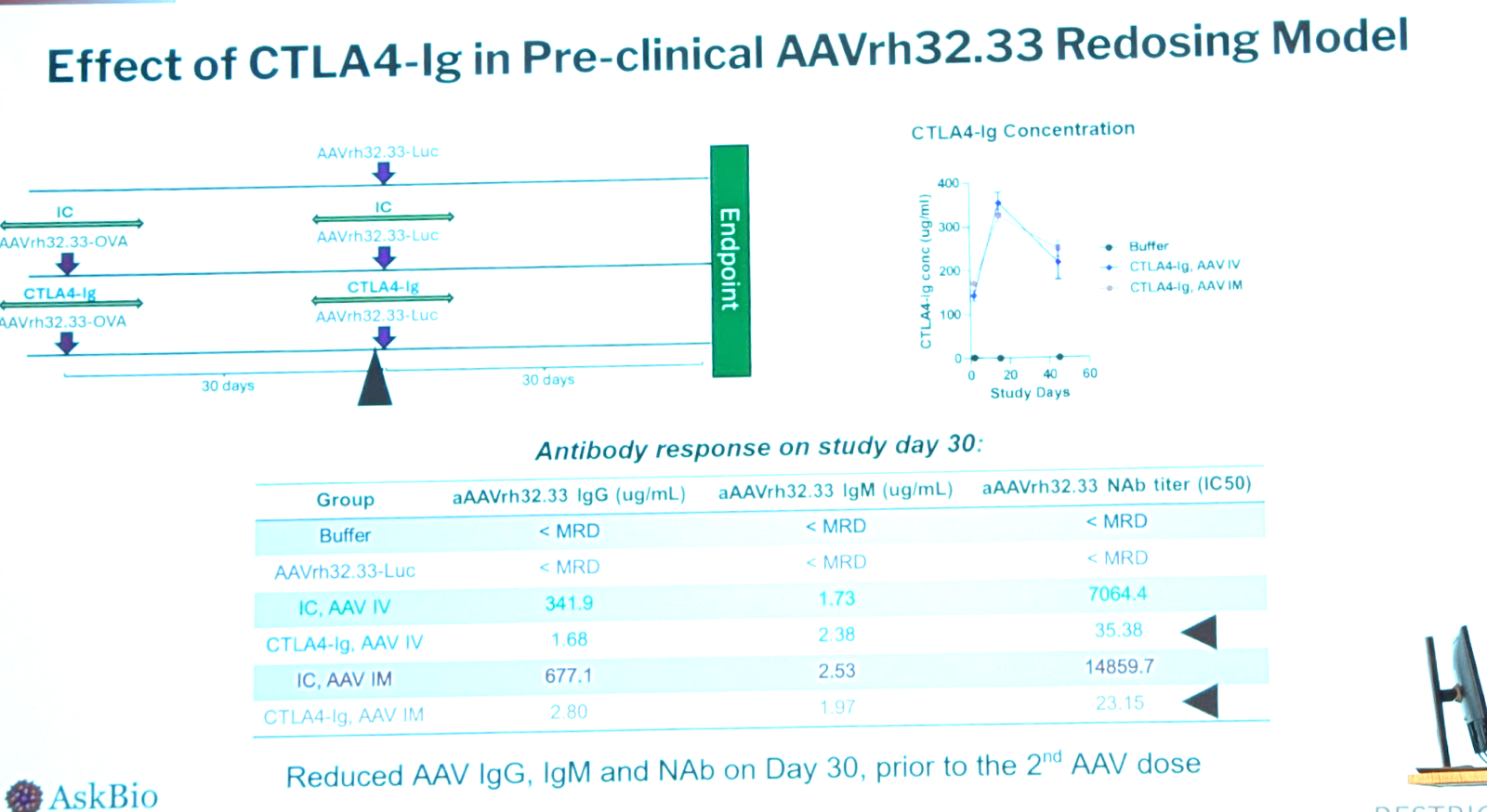
Another goal is to suppress the developing immune response upon AAV administration. Corticosteroids are the most used, but people have also used T cell inhibitors such as tacrolimus, B cell inhibitors/depleters/signaling blockers. Rituximab and rapamycin are both widely used; AskBio used those two as a “standard of care” and compared an alternative approach of inhibiting T cell activation using CTLA4-Ig. There are 4 approved versions of CTLA4-Ig including abatacept (for rheumatoid arthritis) and belatacept (for kidney transplant). In their mouse model, CTLA4-Ig works better than rapamycin + anti-CD20. In the immunogenic mouse model the CTLA4-Ig monotherapy durably blunted the T cell response. Rapamycin + anti-CD20 helped but not as much. They replicated this in a small NHP study with N=3 per group.
Could CTLA4-Ig make it possible to give a second dose? They gave mice AAVrh32.33 expressing OVA, and then another dose 30 days later expressing luciferase. They compared with and without the OVA dose, with and without CTLA4-Ig, and by intravenous and intramuscular routes. The CTLA4-Ig was remarkably effective at reducing IgG response but not effective against IgM; use of CTLA4-Ig permitted a second dose of AAV intramuscular, but not intravenous — for intravenous AAV, zero transgene expression was seen after the second dose, even with CTLA4-Ig treatment. Data below:
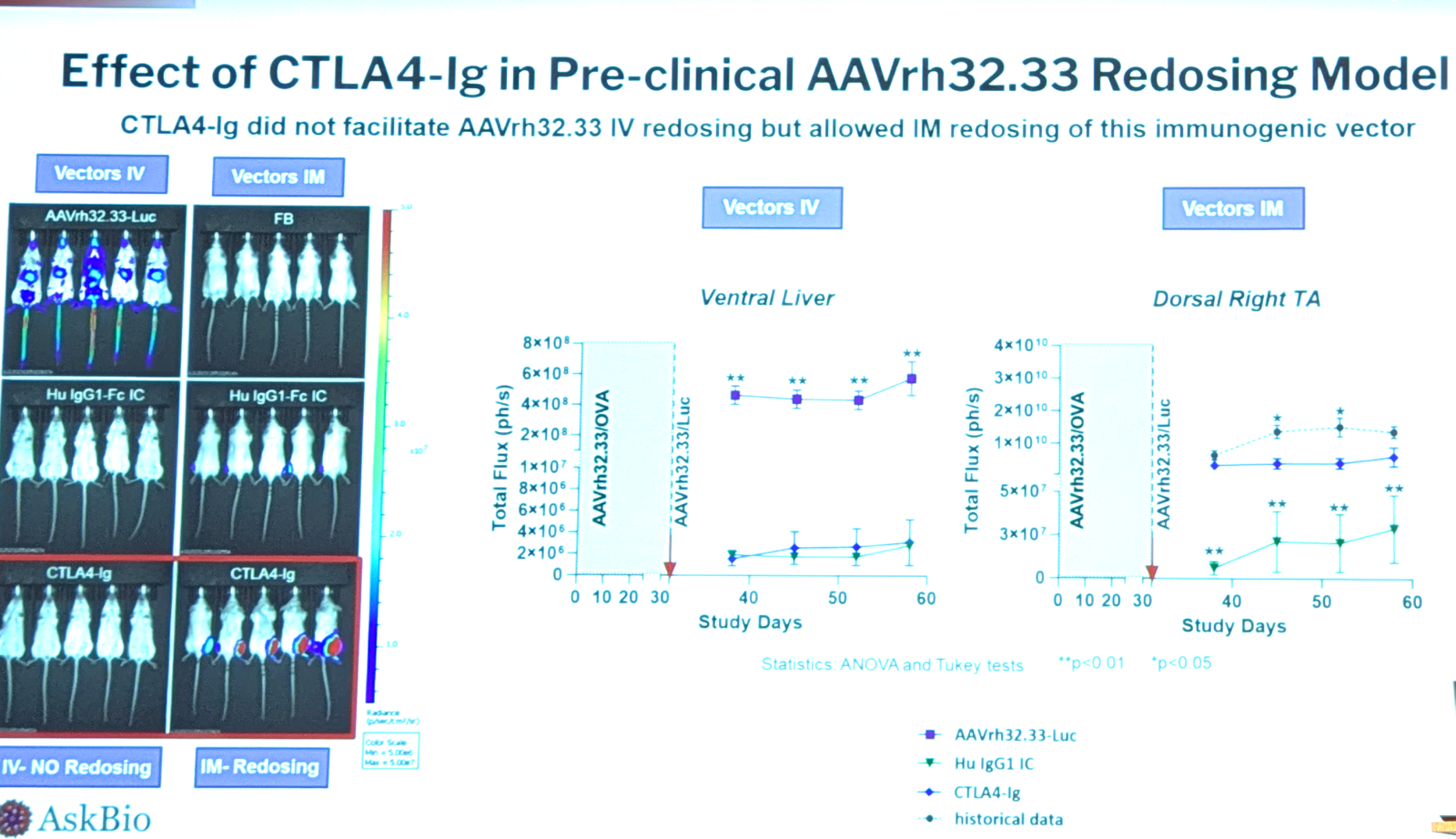
AAV-specific antibodies modulate immune responses to AAV | Klaudia Kuranda, formerly Spark Therapeutics
A large recent survey of AAV pre-existing immunity found that seropositivity differs by capsid and increases with age [Wang 2024].
Clinical trials have demonstrated cases of thrombotic microangiopathy (TMA), a potential safety concern. Pre-existing NAbs increase cytokine response and complement activation after AAV administration [Zaiss 2008]. EGTA can block classical complement pathway. Depletion of anti-AAV IgG prevents complement activation [West 2023]. Cleaving both IgG and IgM is better than just one or the other [Smith 2024]. People with pre-existing immunity have responses to AAV therapy that are distinct from those in seronegative patients [Kuranda 2018]. In hemophilia AAV5 trials (etranacogene), they do see elevation of liver enzymes such as ALT, but it was not worse in patients with detectable NAbs [Pipe 2023]. This may be because there is some in vitro evidence that AAV5 NAbs are not actually very neutralizing and have lower affinity than NAbs against other serotypes. But there are also two countervailing forces here. NAbs can promote complement, cytokine, and increased Ag uptake into APCs, but they also cause AAV neutralization and clearance, which is expected to reduce transduction and thus ameliorate liver toxicity.
To date, the evidence for save AAV gene therapy administration in seropositive patients is limited to AAV5 and one patient with AAV2. (*I think this refers to [Pipe 2023] and [Manno 2006] respectively).
Overview of AAV Antibody Assay Development & Efforts Toward Standardization | Brian Long, 4D Molecular Therapeutics (4DMT)
3 aspects of AAV determine its immunogenicity:
- Capsid
- Transgene cargo
- Innate sensing of nucleic acids and pathogen-associated molecular patterns (PAMPs).
Dr. Long’s focus for today is capsid. There are two AAV antibody assay formats:
- Cell-based neutralization (NAb) or transduction inhibition (TI) [Falese 2017]. You test the ability of plasma to block transduction of cultured cells by an AAV5-GFP or AAV-luciferase reporter vector. Signal is inversely proportional to the antibody concentration or titer.
- ELISA-based binding antibody (BAb or TAb). This measures antibodies binding to the capsid. This is a sandwich ELISA format. You have to serially dilute plasma until signal goes away.
- BAb: You bind the capsid onto the plate surface, let anti-capsid antibodies from plasma bind to it, then use a detection antibody with a fluorescent substrate to read it out. You can use isotype-specific detection antibodies, so you only measure IgM or only IgG.
- TAb: measures divergent isotypes. You use anti-kappa or anti-light chain antibodies which are agnostic to Ig isotype.
NAbs block binding of the AAV to its receptor on the target cell. BAbs may not neutralize the AAV, but they do enhance its clearance via complement and FCy receptors on phagocytes, and can dramatically change biodistribution, sending it to the spleen.
TAb and NAb assays do not always agree with each other, see an exanmple for Factor IX [Majowicz 2019]. Some of this may just be the different sensitivity of GFP versus luciferase. In some trials, they excluded patients who had either BAbs or NAbs, and included only double negative patients. Is that the right decision or do only BAbs or only NAbs matter? Dr. Long did an NHP study to try to answer this at Biomarin; Factor VIII expression from an AAV5 was only negatively impacted when the animals were both BAb and NAb positive. Biomarin changed its trial inclusion criteria based on these findings.
Why aren’t these assays more standardized? Actually there are multiple guidances and standard protocol papers [Shankar 2008, Gorovits 2020, Gorovits 2021, Braun 2024]. What’s less standard is where do you draw the cutoff - how positive of a titer merits exclusion. At two extremes: Biomarin for hemophilia A excluded all Ab-positive individuals, while etranacogene in hemophilia B accepted all comers and found a lack of efficacy only at very high titers.
For Biomarin’s valoctogene, the pivotal Phase III excluded AAV5 TAb+ individuals. FDA approved a companion diagnostic, and the label for valoctogene requires seronegativity. However, in that Phase III, there were 3 individuals who screened negative for TAb but later turned out to have been positive, and 11 individuals who screened negative for NAb but turned out to have been positive. In all cases, these people still had reasonable Factor VIII expression, albeit slightly lower than the totally negative people [Long 2024].
Applying learnings from the SARS-CoV-2 pandemic to assessing AAV seropositivity | Tomas Baldwin, University College London
Dr. Baldwin’s uses mass spec to try to overcome known problems with measuring immune response to AAV [Schulz 2023]. His assay multiplexedly measure immune complexes against patient serum. They can measure many different IgG and complement in one shot, in about 3 hours time. They tested this a lot on SARS-CoV-2 and it correlates well with the gold standard Roche assay. They were ready to roll it out in the UK, but then the UK stopped testing for COVID. They then pivoted to AAV. They validated their platform for AAV and then tested it in a cohort of N=15 pre- and post-onasemnogene SMA patients. 58% were IgG1-positive pre-therapy. IgG1 went up massively after onasemnogene treatment in 100% of patients, continued to rise through 8 months post-infusion, and correlated with a strong complement response across the entire complement pathway. IgG2 rose only slightly after treatment, and only in the already-seropositive people. IgG4 had pre-existing binding in some patients but did not rise after treatment.
Can the antibody or complement response predict severe adverse events? They have only 2 paired pre/post samples. Patient 1 had some IgG4 but no IgG1 or 2 and had a benign response. Patient 2 had IgG1 and IgG2 and had a severe reaction. It’s still just N=2 but they are interested in learning if this is predictive.
They can also use their assay to assess cross-reactivity between AAV8 and 9.
Anti-AAV Seroprevalence in a DMD Cohort & Assessment of Impact on Efficacy Preclinically | Sharon McGonigle, Solid Biosciences
Solid is developing SGT-003, an AAV-vectored microdystrophin for Duchenne. It uses AAV-SLB101 which is a muscle-tropic novel engineered capsid based on AAV9 but with a 7 amino acid insertion. It provides improved biodistribution and transduction in muscle and reduces liver transduction. It encodes a unique microdystrophin gene. The FIH study called Inspire Duchenne (NCT06138639) launched in 2024 and 3 patients have been dosed at 1e14 vg/kg. Biomarker and 90-day biopsy expression data on first 3 patients will be released in Q1 2025.
The trial requires that patients be “Negative for AAV antibodies”. The sponsor’s main concern is that the therapy wouldn’t be effective in patients with antibodies. They tested serum in a cohort of 58 Duchenne patients from Cure Duchenne Link, using the TAb and NAb assays that Brian Long described earlier. They found that TAb and NAb titers were very tightly correlated both for wild-type AAV9 and for AAV-SLB101:
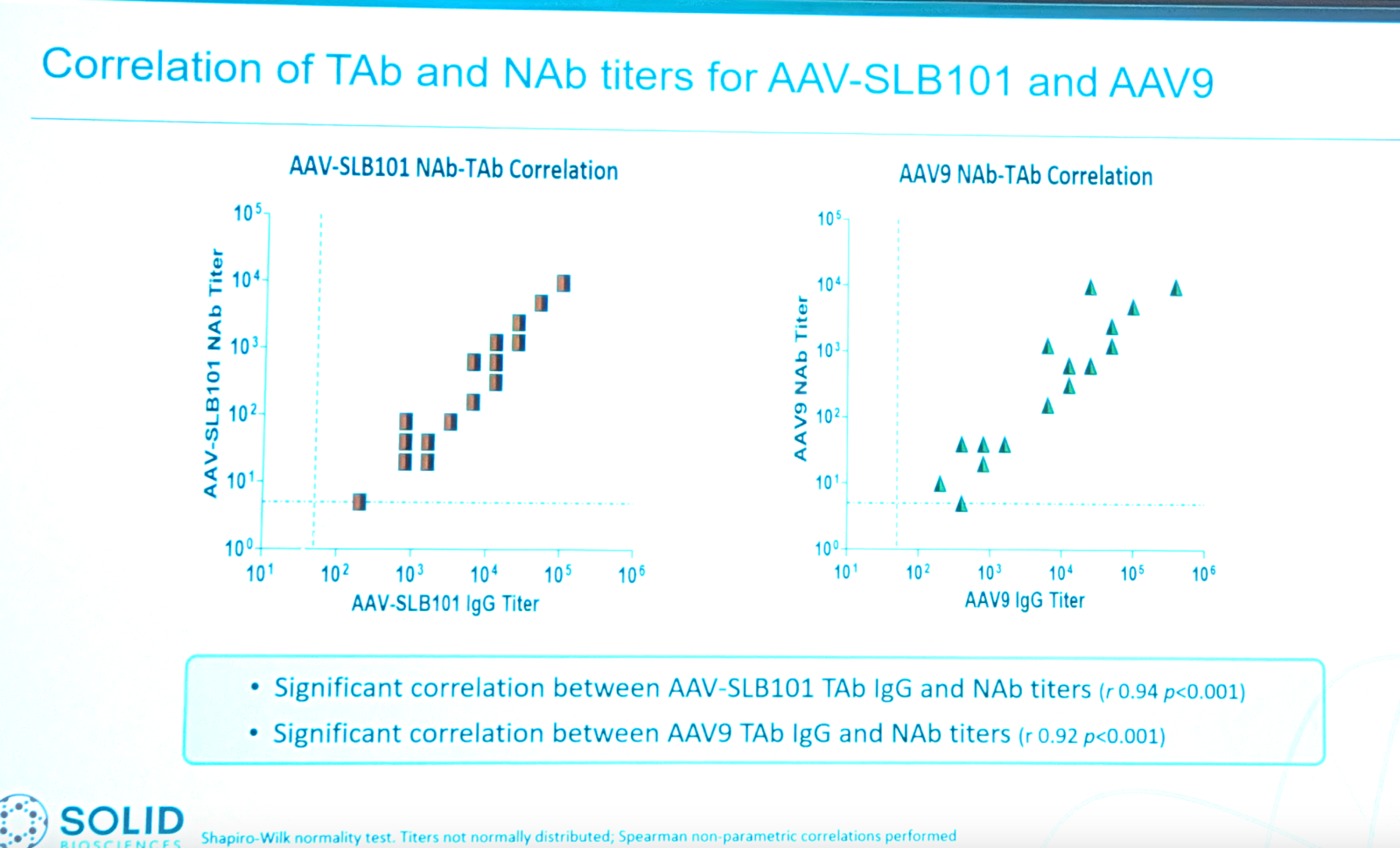
Because TAb and NAb are correlated, she’ll only talk about TAb for the rest of the talk. TAb titers were very tightly correlated across AAV9, AAV-SLB101, and AAV-rh74. Many patients had very high titers, even above 1:6400. Where do they want to draw the inclusion cutoff to maximize inclusion without sacrificing efficacy? They did a mouse study to figure out the titer where they actually lose transgene expression, using a luciferase reporter AAV-SLB101 CK8-LUC with full-body IVIS as the readout. They found that liver gets plenty of AAV even at high TAb titers, but diaphragm and skeletal muscle can lose transduction even at low TAb titers.
While preclinical studies can help decide a cutoff, there will always be some patients above that cutoff. They are very interested in IgG depleting or degrading strategies, immunomodulation, and so on to allow them to address more patients.
Abs to AAV: durability, cross reactivity, & window for dosing | Roberto Calcedo, Affinia Therapeutics
Prevalence of AAV seropositivity is pretty much the same anywhere in the world and in healthy vs. patient populations [Calcedo 2009, Verma 2023]. Newborns get anti-AAV antibodies from their mothers but the titer declines in the months after birth [Calcedo 2011].
Seropositivity is high for a wide range of natural AAVs. Only 2-3% of people are seropositive for AAVrh32.33, but no one wants to use this because it’s extremely immunogenic. They studied the natural history of AAV immunity in a social population of chimpanzees [Calcedo & Wilson 2016], and found that once immunity to AAV8 is acquired there is generally some cross-reactivity to other serotypes. In humans, too, cross-reactivity is prevalent, though people may have higher titer for one type than for another. In an intramuscular AAV1 AAT gene therapy trial, they saw that patients developed responses not only to AAV1 but to AAV2, 8, and 3B as well [Mueller 2013]. In NHPs (cynomolgus and rhesus) dosed with 3e12 vg/kg intravenous AAV8, even modest NAb titers blocked intravenous AAV and shifted biodistribution away from liver and towards spleen [Wang 2011]. But note this is dose-dependent and you may be able to overcome it at higher doses. When titers are low, NAb and TAb are not well correlated. For intramuscular delivery, moderate NAb titer does not affect potency; only at 1:320 do you get reduced transduction. Same for intrathecal administration in Giant Axonal Neuropathy (GAN) [Bharucha-Goebel 2024].
Studying the impact of AAV seropositivity in nonhuman primate models | Juliette Hordeaux, Gemma Therapeutics
NHP are generally a good model for studying AAV gene therapy immunogenicity, although because they have amino acid differences versus human transgenes, they are in some cases more likely to have adaptive immune responses to the transgene. Seropositivity prevalence to AAV9 is similar in momkey as in human.
They split a group of cynomolgus macaques into those with and without pre-existing antibodies (NAb titer > or < 1:5). They dosed 5e13 vg/kg AAVhu68 with a therapeutic transgene under the CAG promoter [Hordeaux 2023]. The ALT elevation was actually worse in the animals without pre-existing antibodies. But the NAbs, by preventing transduction, also prevented liver toxicity. They also tested injecting 3e13 vg per animal intra-cisterna magna (ICM). They used an FcRn antibody to counter pre-existing NAbs and were able to achieve cardiac transduction with systemic administration [Horiuchu 2023].
Overal conclusion, in all their NHP models they never saw a safety issue with pre-existing antibody titers. The only issue is efficacy.
Intrathecal Gene Therapy for Giant Axonal Neuropathy: Immunological Considerations & Observations | Carsten Bonnemann, NIH
Giant axonal neuropathy (GAN) is caused by loss of function of gigaxonin (GAN), an E3 ubiquitin ligase adaptor required for breakdown of intermediate filaments. All different intermediate filaments such as GFAP, NfL, vimention, and even keratin in hair are affected.
They believed GAN was a perfect test case so they did the first ever intrathecal AAV therapy [Bharucha-Goebel 2024]. The therapy was scAAV9 JeT-hGAN, using a weak but ubiquitous promoter. The trial (NCT02362438) escalated from 3.5e13 vg to 1.2e14, 1.8e14, and 3.5e14, always in a 10.5 mL dose volume. Patients were in the Trendelenburg position to try to maximize distribution to brain. Patients were symptomatic, age 6-14 years, with clear electrophysiological changes in CMAP. There was low systemic exposure. They did not exclude people with pre-existing NAb. At baseline about half were seronegative, a quarter positive and a quarter borderline. The antibody responses to the therapy were possibly just slightly faster in the seropositive people, but plateaued at the same level regardless.
At the high dose level, all patients also received rapamycin/sirolimus, which may be why the high dose individuals had longer persistence of detectable vector in blood post-dosing. Some patients had homozygous or compound het LoF, in which case they are immune naive to the transgene (“CRIM Neg”), while others had missense and would be expected to be more tolerant of the transgene (“CRIM Pos”). All CRIM Neg patients had to go on immunomodulatory therapy with tacrolimus and rapamycin, and prednisone at high dose level. CRIM Pos get just rapamycin, and prednisone at the high dose.
One more advanced patient passed away of disease complications and the family donated their body for autopsy. The biodistribution across the brain was reasonably consistent with what was expected based on preclinical work in NHP [Gray 2013].
The run-in natural history of patients leading up to their enrollment in the trial was consistent with natural history [Bharucha-Goebel 2021].
They argued based on a Bayesian analysis that was a 90+% posterior probability that the top 3 out of 4 dose levels had slowed the slope of decline, and therefore consider the trial to have met its endpoint.
There was no evidence for DRG tox, and indeed, clinical sensory endpoints moved in a positive direction suggesting there was rescue of DRGs by the therapy.
Toward AAV gene therapy in seropositive Crigler-Najjar patients | Giuseppe Ronzitti, INSERM / Genethon
Crigler-Najjar syndrome is a deficiency of UGT1A1 in hepatocytes which if untreated causes severe neurotoxicity and death. Prevalence is 1 in 1 million births. The standard of care is phototherapy, in which patients must be exposed to blue light 10 - 14 hours every day, which allows bilirubin to be converted to other molecules that can be broken down. The only cure is liver transplant but then the patients require lifelong immunosuppression and really, having had a liver transplant is just a different disease.
They did a trial of intravenous AAV8 encoding UGT1A1 at 2e12 to 5e12 vg/kg [D’Antiga 2023]. Even with phototherapy the patients had bilirubin of 250-400 μmol/L at baseline. Within ~1 week it went down to <100 in 100% of participants. The trial met its primary efficacy endpoint and was stopped at 16-17 weeks. Blinding was impossible because the therapy is so effective that patients who are yellow in skin and eye color due to bilirubin turned back to a normal skin color after treatment. They are still going to do a pivotal trial though.
To manage seropositivity, they wanted to use an IgG degrading enzyme called “immunoglobulin G-degrading enzyme of S. pyogenes” (IdeS), which was clinically validated in transplant patients [Jordan 2017] and is now approved in Europe.
They did a bunch of preclinical studies (Leborgne unpublished). In cynomolgus macaques (N=2 per group) they tested whether IdeS treatment 2 mg/kg at 2 days pre-AAV could improve efficacy of 2.5e13 vg/kg AAV9. It gave a slight increase in VGCN per diploid genome and a larger increase in transgene mRNA expression. Then they used NZW rabbits to do an AAV re-dose study. They dosed AAV8 vector 5e12 vg/kg, then waited ~2 months, then dosed IdeS 1 mg/kg then 2 days later dosed AAV8-hFIX 1e13 vg/kg. IdeS resulted in a >10-fold decrease in anti-AAV8 NAb titers. They are now announcing a new trial which will be open to seropositive patients who were excluded from the first trial. And they are hoping that eventually this will allow re-dosing.
AAV Redosing | Barry Byrne, University of Florida
Hours after a 1e13 to 1e14 vg/kg dose of AAV systemically, the virus concentration is 1e11 per mL in blood. That high initial exposure injures endothelia and contributes to innate immunity, which kicks in within hours of dose while adaptive immunity takes days or weeks [Mendell 2022]. Pre-existing antibodies do contribute, but no matter how high the pre-existing titer, these antibodies are undetectable at 1 hour post-dose because they all get saturated by the AAV and then cleared.
No assay currently differentiates seronegative from “true naive”. Only an IgM assay post-exposure can figure out if a person was truly naive. Either TAb or NAb assays are difficult to standardize, so it is easier to use an external control approach.
Last month was the 25th anniversary of the first patient receiving a systemic AAV (an AAV1 in a muscular dystrophy trial). That first patient is still alive and still seropositive. Seropositivity appears to never wane.
TLR9 is the main detector for CpG islands in nucleic acids. Even though we produce AAV in eukaryotic cells, there are not enough replication rounds in eukaryotic cells to get rid of the CpG signature from the prokaryotic cells we used to produce the plasmid. In response some people have tried to reduce CpG content in the transgene but this often results in a decline in expression level.
First wave immunity is driven by cell surface receptors, but wherever the vector goes, the antibody goes, and so we do see intracellular sensors turn on too, including the proteasome as well as cytoplasmic and intranuclear viral sensors.
They are studying an AAV therapy in Pompe disease, and many patients are seropositive. Rituximab can deplete B cells, sirolimus can control reg T-cell promotion and expansion and downregulate cytotoxic T-cells. Neither therapy alone is enough. Together they do well at reducing immune response [Corti 2014]. Corti 2017. The first systemic gene therapy patient was for Canavan disease and had been on rituximab and still does not have antibodies against AAV (?).
Based on mouse and NHP data, they launched the first-ever AAV redosing trial in Pompe disease (NCT02240407).
The activation of complement pathway after AAV therapy is terribly complex:
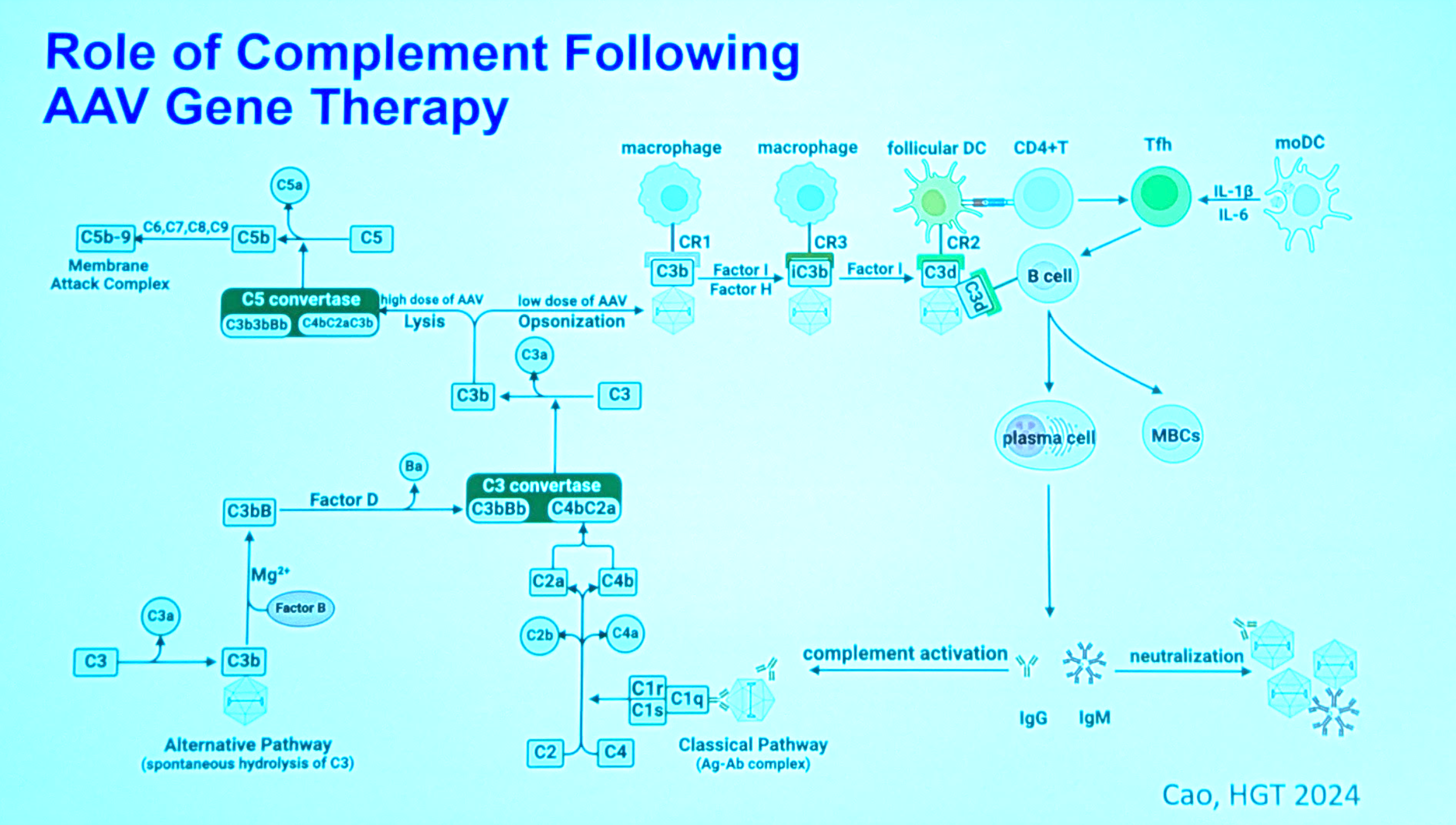
The upshot is that it’s easier to just block the antibodies, as the crucial chokepoint, rather than going after complement itself. They did an entirely clinical study of how immunosuppressive regimens affect complement response in patients receiving AAV therapy [Salabarria 2024].
In some cases they have to use plasmapheresis as a rescue therapy - this happened with one Duchenne patient with hemodynamic instability. Luckily that child recovered. The downside is plasmapheresis does also remove some of the drug and thus reduces efficacy.
They have submitted an IND together with Muscular Dystrophy Association (MDA) to do AAV therapy in two groups of patients, 1 group with pre-existing natural immunity and 1 group with prior exposure to AAV therapy.
They are also exploring a novel approach to manage pre-existing immunity, called MODE, which leverages ASGPR.
Pretreatment of cynomolgus macaques with prednisolone, rapamycin, & rituximab inhibits capsid-specific humoral immune responses to high-dose AAV gene therapy | Barbara Sullivan, Ultragenyx
Today’s talk will focus on two detailed NHP studies.
They hypothesize that thrombotic microangiopathy (TMA) is mediated by anti-capsid immune complexes and IgM activating complement [Muhuri 2021]. This would not be a problem if the patient is seronegative or if the dose is low, because vector is already taken up and cleared from circulation by the time the response is mounted. But in seropositive or high dose patients, there is still significant capsid in circulation when the response is fully mounted.
Their 3-drug approach: prednisolone to reduce inflammation, rituximab to deplete mature naive and activated B cells, and rapamycin to reduce differentiation ti plasma cells. The TMA window is 21 days post-dose, and Dr. Byrne’s work (above) showed that it depends on anti-capsid IgM [Salabarria 2024].
In NHP they gave “PRR” (prednisolone/rituximab/rapamycin) for 29 days before a 2e14 vg/kg AAVhu37-CK8-μDys intravenous dose. The rituximab dose of 10 mg/kg was based on preclinical data from Genentech. The 2mg/kg rapamycin dose is “the cancer dose”. In humans probably both doses would be lower. Both the AAV and the PRR were well-tolerated. PRR yielded a 75%-100% reduction in B cells. Anti-capsid IgM were reduced in the PRR group compared to the “no immunomodulation” group. But in order to see any reduction in anti-capsid IgM and IgG titer, you need to get very very close to 100% depletion. Even 75% or 81% does nothing. You need like 99% depletion.
PRR yielded a 1,000-fold increase in serum AAV, but uptake into tissues was only 2x higher in liver and not detectably better in any other tissue.
Immunomodulation strategies for overcoming antibody responses to AAV | Nicholas Giovannone, Regeneron
In AAV-naive individuals, the question is can we immunosuppress the individuals at their first dose to keep them immune naive and preserve the possibility of re-dosing. In seropositive individuals the qeustion is how to supress their NAbs to enable treatment.
To prevent initial immune response, the CD40 TNF superfamily receptor is crucial for B cell activation by T cells and promots T cell activation via DC licensing. In the germinal center it is critical for regulating B cell affinity maturation, class switch, and differentiation. The ligand of CD40 has been targeted before including by people in this audience. Regeneron has taken a different approach and made fully human effectorless IgG antibodies against CD40 itself. The goal is to keep naive individuals naive and enable re-dosing.
They use CD40 humanized mice. They prophylactically dose 40 mg/kg anti-CD40, then dose AAV8 6.6e12 vg/kg the next day, then continue to dose the anti-CD40 2x/week for 4 weeks, then dose 6.6e12 vg/kg AAV8 again, this time encoding GFP. The second dose achieved transduction comparable to animals that had never seen the first dose. It both ablates responses and suppresses germinal centers. Anti-capsid specific T cells by ELISpot were also suppressed.
What about pre-existing immunity? This is a problem because it is harder to kill the cells that are already making the antibodies. Terminally differentiated B cells lack common B cell markers. They are highly secreting and can hang out in bone marrow for literally decades. They are the source of most circulating antibodies. They are the source of long-lived AAV immunity. Anti-CD20 antibodies such as rituximab deplete early B and memory B cells but not plasma cells and so won’t fully suppress antibody secretion. Regeneron wanted to test a strategy of plasma cell depletion. They use bispecific antibodies which express B cell maturation antigen (BCMA) and CD3 which is expressed on T cells. Bringing them together induces cytotoxic killing of plasma cells by the T cells. As early B turn into mature B and then plasma cells, BCMA is upregulated at precisely the stage where rituximab stops working. Regeneron made BCMA- and CD3- double humanized mice. They had controls and 3 experimental arms: BCMAxCD3 to kill plasma cells, anti-CD19/CD20 to deplete B cells, and efgartigimod to accelerate IgG clearance. (The reason why you need efgartigimod is that the clearance of existing IgG is too slow, 7 days in mice and 21 days in human. You need FcRn blockade to get them degraded.) Animals got AAV first dose at day 0 to simulate pre-existing immunity, then got immunosuppressive regimens beginning day 70 or so. None of the monotherapies was helpful. BCMAxCD3 and efgartigimod in combination brought titer down by a log or two. All 3 therapies in combination brought antibody titers down to undetectable. Why did we need all 3? It’s possible that because we only waited 70 days there is still primary B cell response that we need to kill B cells in order to suppress. But it also turns out that efgartigimod is highly immunogenic in mice and they develop anti-drug antibodies which not only clear the efgartigimod but also are cross-reactive against BCMAxCD3, thus totally tanking the efficacy of the therapy. The triple therapy also restored transduction of a 2nd AAV dose to a level equal to the 1st dose, whereas any monotherapy didn’t work and the dual therapy only partially restored.
Panel Discussion + Q&A with Session 3 speakers
There are multiple reasons to suppress initial immune response. One is to be able to re-dose years from now if response wanes — less a risk for neurons but an issue for muscle. Another is to be able to split doses over multiple smaller doses because we are saturating cellular receptors in the large doses we give. A third is to be able to do within-subject dose escalation over a 3-month or 6-month period. We need to socialize this with the agency. And the bar for enabling re-dosing in a 3- or 6-month window may be lower than “remote” re-dosing. Anti-CD40s may be a key part of enabling this.
In the expanded access study for Canavan’s, the intended window of coverage was 6 months but we saw persistence of vector genomes in blood longer than we expected, still strong at 6 months. So we gave a maintenance dose of rituximab. The child was going back to daycare and worried about opportunistic infection, so we added IVIG. The IVIG quickly cleared the vector from blood and the child remains seronegative.
Ben Deverman likes the idea of if you have a targeted AAV that transduces within 1 day, could you dose with IVIG afterwards to clear out the residual AAV so that it’s not still there for the patient to develop immunity against? The panel likes this idea.
There is a temptation to operationalize the immunosuppressive regimen and do what’s easy, such as a single dose of rituximab, but that wouldn’t work. For rheumatoid arthritis the total 1500 mg dose can be divided in 3, delivered at time minus 2 weeks, minus 1 weeks, and right before dosing, to catch all the emerging pre-B cells that are continuing to mature over the time leading up to the AAV dose. You cannot shorten this regimen because that is just how long B cells take to mature. It will keep the B cells depleted for at least 4 months, maybe 6.
Has anyone tried anti-CD19 antibodies in humans for AAV? Nope. And it can’t be done in NHP because they are all non-cross-reactive.
Engineering receptor-targeted antibody-evading capsids for CNS gene therapy | Ben Deverman, Broad Institute
Dr. Deverman focuses on engineering AAVs to use the vasculature to reach all regions of the CNS. His efforts to engineer AAVs to cross the blood-brain barrier have focused on known human receptors, chiefly transferrin receptor (TfR1). Their new vector, BI-hTfR1, has achieved widespread delivery [Huang & Chan 2024]. They have now generated 2nd generation versions where they can achieve that same high level of transduction with 2e12 vg/kg, which is 50-fold lower than the FDA-approved doses for onasemnogene and delandistrogene.
Their other challenge is to engineer in antibody-evading technology to enable AAV to address a larger percentage of the patient population. They made a library of >1e5 capsid variants and screened them for ability to evade serum antibodies, still bind TfR, and maintain manufacturability. First they screened a panel of human serum samples in dilution series to quantify half-inhibitory dilutions (IC50), with a limit of detection (LOD) of 1:4. They then screened 50 capsids at a single dilution against 8 human serum samples, starting with sera that had neutralization above LOD but still low. They picked 5 capsid variants that appeared to have improved evasion, and tested them against a full panel of sera. They then confirmed in vivo that these 5 improved capsids have similar transduction efficiencies as the parent. Based on all available data they narrowed it down to 1 best candidate. They tested this 1 candidate, versus AAV9, AAV5, and the original engineered capsid. There were some serum samples where the new capsid evaded immunity completely, and others where it changed what dilution was required to see neutralization.
The antibody-evading scaffolds can also be scaffolds for other tropism-enhancing insertions. They wanted to test whether they could also do this for Sharif Tabebordbar’s MyoAAV capsid to retain both engineered properties. Sure enough, they found a different antibody-evading amino acid sequence that was compatible with MyoAAV, evading antibodies while preserving the enhanced muscle uptake and decreased liver transduction.
T-cells: Friend or Foe in Gene Therapy? | Allison Keeler, UMass Worcester
Today we’ve heard a lot about cytotoxic T cells but not about T regulatory cells (Tregs, pronounced T-regs), the T cells associated with immune tolerance, which are our friends.
In some trials such as hemophilia, they observed T cells stimulated by capsid to produce interferon gamma [Nathwani 2011, Rangarajan 2017]. They managed this with corticosteroids. But there was also an alpha-1 antitrypsin trial cited earlier [Mueller 2013] where they did no immunosuppression and they did see CD8 cells but they also saw Tregs. And 2 patients even had pre-existing immunity. But this was intramuscular so the immune response is very different.
While many trials pre-screen for humoral immunity as an exclusion criterion, no one is screening for cellular immunity. Even prevalence is not as well documented. One recent study did both IgG and ELISpot screening on donors [Xicluna 2024]. They found that there were individuals who were IgG-negative but not truly naive, because they had ELIspot positivity. It’s been suggested that the pre-existing T cell response is primarily CD8-mediated.
How would pre-existing immunity influence T cell response to AAV gene therapy? Surprisingly in HOPE-B [Coppens 2024] there was actually less T cell response in patients with pre-existing NAbs. Dr. Keeler’s lab is working on engineering AAV-directed Tregs [Arjomandnejad 2021]. T cell response can cause loss of transgene expression and they showed in a mouse model that the engineered Tregs preserve transgene expression.
Fireside chat with Jim Wilson (Gemma Therapeutics) & Peter Marks (FDA)
Peter Marks said that FDA CBER is “leaning in” to AAV gene therapies and wishes they could see dozens per year. They view rare diseases as a great proving ground to get AAV gene therapy right. So many diseases are potentially approachable by AAV but no one is going to make AAV for a common disease yet. He spoke about 3 FDA CBER initiatives to improve regulatory pathways for AAV gene therapy:
- Continuous communication. The traditional way FDA communicates is that sponsors request a type A, B, C, or D meeting and then wait 30 - 75 days to have one scheduled. A lot of wall time is wasted just waiting. One lesson learned from Operation Warp Speed for SARS-CoV-2 vaccines was that continuous feedback and conversation with sponsors can accelerate things a lot. The model is that sponsors ask questions to a Regulatory Project Manager who interface with the team (FDA scientists) and if necessary, set up a call. This will require a lot more resources — meaning more staff time — so if they’re going to do it more broadly they need data to show that it actually improves things. Accordingly right now they are in a pilot with 4 programs in CBER and 3 in CDER, and gathering data on whether this continuous communication model reduces the frequency of clinical holds and other quantifiable milestones. If they can convince themselves that it shortens development time they will expand the pilot.
- Using Accelerated Approval more. We could debate how appropriate Accelerated Approval is in common diseases, but for rare diseases, it is clearly critical to getting therapies to patients in need in a timely fashion. Moreover, it ensures there is still more oversight on safety than you have in a Right To Try scenario. It is still very rigorous and scientific, while being more feasible and appropriate to disease biology.
- A pilot with EMA to enable a single joint submission to both agencies and pre-agreed criteria for evaluation at both agencies. The hope is there will be fewer gaps where a therapy approved in the U.S. is still unavailable to E.U. patients. Moreover, by doubling the market size with minimal additional effort on the sponsor’s part, it will incentivize more investment in drug development.
Scientists at CBER are transitioning to a new framework for statistical evaluation of drugs. The old model was based on frequentist statistics and having very strict statistical cutoffs and numbers of trials. The new model is more of a flexible “totality of the evidence” approach. It is still statistically rigorous, but the statistical models can be Bayesian. This will mean we occasionally have products that get Accelerated Approval and later have to be pulled from market because they fail confirmatory trials. Hopefully that will be rare.
He also talked about strategies for engaging with FDA. He said if you have a disagreement with FDA, the first thing to check is whether you have a common understanding of the scientific evidence. Often disagreements are because the two sides are not looking at the same dataset. It’s like marriage counseling - you want to make sure that each party is hearing the other one. So it’s good after you present evidence to ask FDA to mirror it back to you to see if they understood the key messages and both sides are on the same page about what the evidence says. If ultimately you feel you’re getting advice that doesn’t make sense scientifically, you can ask for supervisory review, meaning, escalating to the next higher level of review. As long as it is done politely, it’s not bad for the relationship.
After being profoundly short-staffed a couple years ago, where is FDA today with regards to staffing and responsiveness? As part of recent PDUFA updates they’ve added 125 new staff to CBER focusing on cell and gene therapy – 100 in the office for cell and gene therapy, and 25 in stats, finance, and inspections. This has reduced wait times for meetings significantly, reduced the number of “written response only” meeting responses, and 50% fewer clinical holds. Also one reason why short-staffing was so severe for a while is that pulling people off of their normal duties for pandemic response was very inefficient. A lot of things done in response to pandemic response — both COVID-19 and at a smaller scale, mpox — were things that can be done very much more efficiently if you do it every day. So they are putting together a specialized staff to respond to emergencies, so that another outbreak would not require pulling people off of their normal duties.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.