We have an open IND. What comes next?
Today, an exciting announcement: we have obtained permission from the U.S. Food and Drug Administration (FDA) to begin a first-in-human clinical trial of a divalent siRNA drug candidate for prion disease. We are not ready to dose patients just yet, but clearance from FDA is a big milestone, and opens the possibility of a human trial in the near future. In this blog post, I’ll recount how we got here, what this means, and what happens next.
If you want to make sure you hear about the next developments, then follow this blog, join our mailing list, and consider joining PrionRegistry if you want to volunteer for research.
the journey to a drug candidate
In 2019, Dr. Anastasia Khvorova of UMass Medical School visited the Broad Institute to speak about her career-long journey to engineer better short interfering RNA or siRNA drugs. She focused on a milestone that was then very recent: her lab’s invention of what they call divalent siRNA [Alterman 2019]. More about the science of this type of drug in a moment.
Sonia and I had already been hearing rumors about this work for months prior to its publication, and were thrilled to see the advance. Up until that time, antisense oligonucleotides (ASOs) were the only game in town for lowering a target protein in the brain. If you follow this blog, you know about the rationale for ASOs in prion disease, and you know that we already had a longstanding collaboration on ASOs with Ionis Pharmaceuticals, which looked promising, and still does — it’s now in a fully enrolled Phase I trial). But we needed another shot on goal. Anastasia’s divalent siRNA publication showed very deep and durable knockdown of a target gene (HTT), and we thought, if we can do this for our gene, PRNP, that would be really promising.
We approached Anastasia about a collaboration, and within a few months we were off to the races. Her team quickly screened siRNA sequences against both mouse Prnp and human PRNP, and filed a patent on several of them [WO2021173984]. Putting together her lab’s preliminary data on siRNA sequences and our lab’s preliminary data on new mouse models with human PRNP and an assay to quantify PrP lowering in the brain, we applied for a NINDS IGNITE grant from the National Institutes of Health (NIH) to support development of a drug. Amazingly, we won it. It was our newly independent lab’s first NIH grant.
UMass sent us their siRNA molecules to test in our lab. Our new postdoc, Juliana Gentile, got to work testing all these siRNAs, first in cells and then in mice, varying both the siRNA sequence and the chemical configuration to find the best molecule. The main goal was to find a really potent siRNA against human PRNP that could serve as a drug candidate. But the mouse Prnp and human PRNP sequences are different enough that not many siRNA sequences can target both, and we also wanted an siRNA against mouse Prnp so that we could test its efficacy in prion-infected mice. (As I wrote regarding our base editing study, we no longer want to do studies in prion-infected humanized mice.) The best siRNA that Juliana could find against mouse Prnp was not actually all that potent — it only lowered PrP in the whole mouse brain by about 50%. But that was good enough to ask the question, does lowering PrP with a divalent siRNA improve survival, just as we had seen for ASOs? These are long-running experiments, where a group of animals has to be infected with prions, and then months later, receive drug at a very specific timepoint. I’ll never forget the day between Christmas and New Year’s 2021, Sonia and the kids were all sick with Omicron, daycare was shut down, and I, the last person standing, spent the whole day alone in a windowless room dosing 28 animals with divalent siRNA. If we had waited until the family recovered, the animals’ disease would have been more advanced and it might have been too late to effectively treat them. The experiment worked: divalent siRNA treatment, even a single dose right after the onset of symptoms, made a big difference in the lifespan of the prion-infected animals.
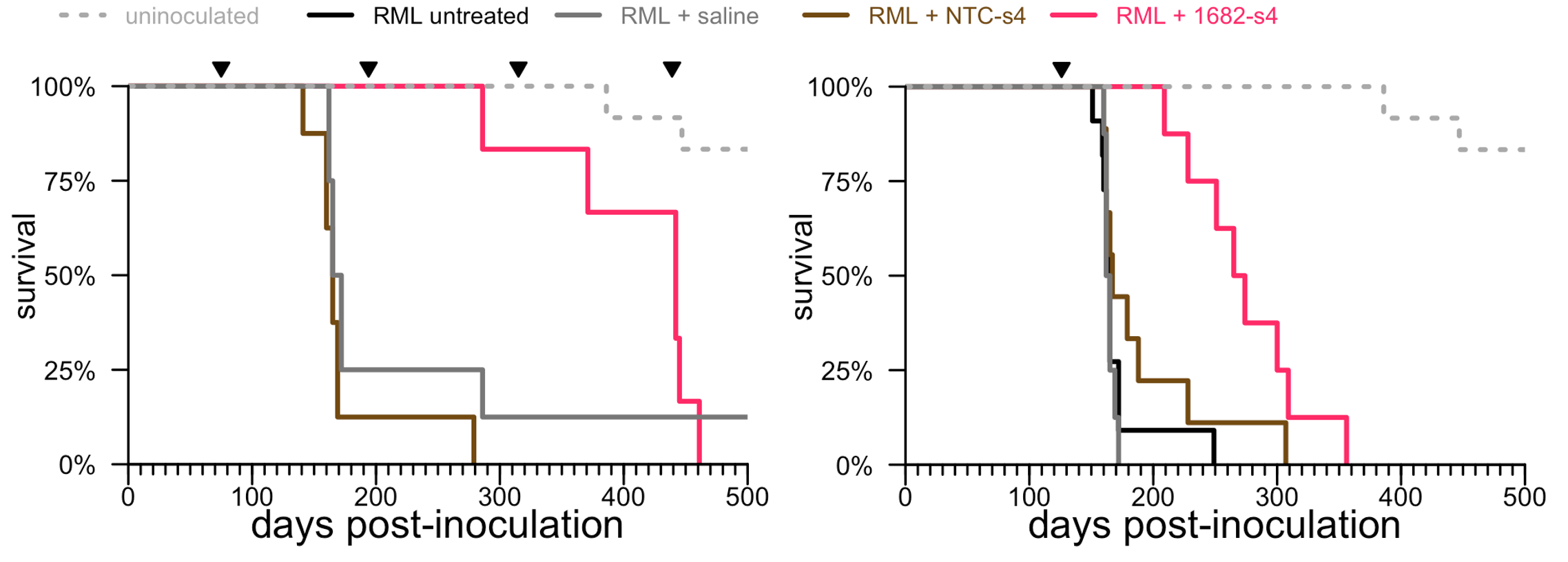
Above: Survival of prion-infected animals treated with a divalent siRNA against mouse Prnp (magenta) versus untreated, saline-treated, and non-targeting siRNA-treated groups (dark colors) and uninfected animals (dashed lines), for both an early chronic treatment paradigm (left) and a single dose after the onset of symptoms (right). Adapted from [Gentile 2024] Figure 2.
Meanwhile, by fall 2022, we had settled on a single divalent siRNA compound against human PRNP which, by every experiment we had been able to do, was the best one of the bunch. We saw a depth of PrP knockdown we had never seen before, with months of durability, and it appeared to be well-tolerated. We desperately wanted this to be another horse in the race to treat prion disease. The choices were stark: either we set out to develop this drug ourselves, or we accepted the grim reality that all this work would be “just a mouse study,” yet another scientific paper that says “this looks promising” but then never actually touches the life of any human being.
Around the same time, we heard of a new NIH funding mechanism, NINDS URGenT, which stands for Ultra-Rare Gene-Based Therapy. Chris Boshoff, a Program Director at NIH, had championed this program as a way out of exactly the predicament we were in: you have promising mouse data, but a first human dose is years of work and millions of dollars away. This program looked to be our only shot, but there was no doubt: even with financial support from NIH to make the task feasible, this was going to be a heavy, heavy lift for us. Sonia and I stood at this crossroads, and after a soul-searching conversation about the pros and cons, the probability of success, and the things we would be sacrificing, we decided to take the plunge. The argument that finally persuaded us: even if we turned out to be wrong and we actually couldn’t develop a drug ourselves, we were going to need to learn that the hard way sooner or later. Given how much we cared, and the fact that at that moment in 2022 there was no clinical trial for prion disease enrolling anywhere on Earth, there was no world in which we were never going to do it ourselves, Sebastian the Crab-style.
interlude: what is a divalent siRNA, anyway?
This section is slightly denser science, and you can skip it if you just want to hear the story.
siRNA stands for short interfering RNA. This type of drug is made up of the same building blocks as DNA and RNA — A, C, G, and T or U — and acts via the RNA interference (RNAi) mechanism. RNAi was discovered in 1998 [Fire 1998], and earned its discoverers the Nobel Prize in Physiology or Medicine in 2006. The siRNA drug is designed to sequence-specifically bind a target RNA molecule in the cell. The siRNA is loaded into the RNA-induced silencing complex (RISC), which uses the siRNA to find the target RNA and then destroy it. The result is less of the target RNA, and therefore, less of the protein that it encodes. An siRNA is therefore a way to lower the amount of one specific disease-causing protein. The first-ever siRNA drug, patisiran for transthyretin amyloidosis, received FDA approval in 2018.
An siRNA is similar to an ASO in that it is an oligonucleotide drug with a sequence designed to bind a target RNA and degrade it. Like an ASO, in humans this siRNA would be administered into cerebrospinal fluid (CSF) by an intrathecal injection, in other words, a lumbar puncture (LP). An siRNA is different from an ASO in that it’s a double stranded (as opposed to single stranded) oligonucleotide, it uses different chemical modification patterns to stabilize it, and it acts through a different cellular pathway (RISC as opposed to RNAse H1).
As of last year, there are 6 FDA-approved siRNA drugs [Jadhav 2024], and tens more in clinical trials. Several different pharmaceutical companies are developing siRNA, each with their own special sauce: different chemical modifications, different conjugates to promote uptake into different tissues in the body, and different sequences for different targets in different disease indications.
A divalent siRNA is a particular special sauce developed by Anastasia Khvorova’s lab [Alterman 2019]. It’s two copies of the same double-stranded siRNA, linked together. Given that the siRNA sequence is the same, why does the molecule need to be double (divalent) and not single (monovalent)? Empirically, studies in mice by the Khvorova lab when they were developing this technology found that single (monovalent) siRNAs of their particular chemistry just did not get taken up and retained in the brain. When the linked two of them together to make a divalent molecule, suddenly a large amount of drug was retained in the brain, taken up into cells, and was pharmacologically active. We still don’t know exactly why, but it probably has to do with avidity, the phenomenon whereby multiple biomolecular binding interactions can be more than the sum of their parts. siRNA drugs need to bind cell surface proteins in order to get internalized into the cell where they can become active, and perhaps the divalent scaffold allows them to do this more efficiently. Many other siRNA drugs for other tisses can be highly active in a monovalent configuration, but for this particular chemical scaffold, in the brain, a divalent configuration appears to unlock a far higher level of biological activity.
climbing the IND mountain
Where I left off a moment ago, in late 2022, we had really promising data in mice. How could we bring this drug to human patients?

In the U.S., and really in every country on Earth, the steps required to get from “looks good in a mouse” to permission to dose a person are not just time-consuming and expensive, they’re also highly specialized studies. There exists a whole industry that, if you don’t work in drug development, you have never seen or thought about, all run by companies whose names you don’t know, specializing in acronyms you have never heard. The chemical synthesis, purification, quality testing, and packaging of the drug — Chemistry, Manufacturing, and Controls (CMC) in industry parlance — has to be done under Good Manufacturing Practices (GMP), which is easily 10 times more expensive than making a drug that is good enough for mice. First you go through all the GMP steps to make a drug substance (DS) — a white powder — then you go through a whole additional process to formulate that powder into a drug product (DP), which could be a pill, or in our case, a vial of sterile injectible liquid. Toxicology studies have to be done under Good Laboratory Practices (GLP), with tens or hundreds of animals, each producing hundreds or even thousands of measurements and pathology slide readings to determine whether the drug is toxic. You analyze the animals’ blood and/or tissues using a GLP-validated pharmacokinetic (PK) assay to confirm how much of the drug is actually there and make sure they got dosed properly. There are drug-drug interaction (DDI) studies, to see whether patients taking any of hundreds of approved drugs should be prohibited from taking your drug. There are genotoxicity studies, to determine whether the drug can mutate DNA and cause cancer or birth defects. All of these have to be done to very exacting standards, with specific documentation practices in place, by experienced personnel with specific training. Doing it in your own academic research lab is out of the question. There’s no way that we could meet all the standards required — it’s not just about doing high-enough quality science, it’s about dotting specific i’s and crossing specific t’s that you’ve never dreamed of.
The goal of all these things is to get you to yet another acronym: the IND, or Investigational New Drug. An IND application is the package of data from all the above studies that a drug’s developer, or sponsor, submits to FDA in order to request permission to dose a human being with a new drug for the first time. To people in the industry, the letters IND, or their European equivalent, the Clinical Trial Application or CTA, have an almost cultish resonance. Job postings ask for people with IND experience; Gantt charts on people’s laptops across the world show elaborate multi-year dependencies all converging on the letters “IND”; biotech startups put out press releases when they have their first IND cleared by FDA, knowing that this milestone will launch them into a new fundraising league with venture capitalists. (Note that we say an IND is cleared, whereas approved is a term reserved for marketing approval, which is what FDA gives you after clinical trials are completed and a drug has been shown safe and effective.)
Dr. Boshoff’s vision for the URGenT program was that NIH would contract out many IND-enabling studies directly — that means all the GMP and GLP and so on, the things required to get you to an IND — as well as giving our lab some direct funding to support these studies, and providing consultants to advise on all aspects of manufacturing, study design, and regulatory filings. Knowing that this level of support was a possibility was incredibly enabling. Without that hope, we could never have taken the plunge. At the same time, from submitting our proposal to NIH in October 2022, we knew it would be the better part of a year before funding could be awarded, plus probably some lag time after that before contracts could be put in place and work could begin in earnest. Wanting to move as fast as possible, we kicked off the drug manufacturing right away using Prion Alliance funds. In June 2023, Sonia and I had our first Pre-IND meeting with FDA, in which we got to ask questions about what would be required for our IND given the particulars of our specific drug and our specific disease. With advice from FDA in hand, we worked with some extremely smart, dedicated, passionate donors to raise funds for some of the other most rate-limiting steps, particularly the first round of toxicology studies and the development of a PK assay, the method for quantifying drug in blood or tissues.
Our financial resources were still quite limited, and we felt that speed was paramount, so we made some difficult tradeoffs. We opted to do a single dose toxicology study in animals, which, FDA was very clear with us, would only permit us to do a single dose trial in patients. We made this decision for several reasons. We could only afford a single dose study, and a single dose study would be faster. Our mouse data showed that a single dose could have activity lasting for 4-6 months, and a single dose at a symptomatic stage of disease extended survival in prion-infected mice by 3.5 months. Thus, considering that many patients with prion disease live only a few months from diagnosis, we felt that even a single dose had the potential to be clinically relevant. Finally, a single dose toxicology study was less risky. As a rule, toxicology studies are already normally done at several times the equivalent of the dose you expect to use in humans; if you add onto that monthly dosing for 9 months (as is typical in chronic tox studies), you’re giving animals a ton of drug, and it becomes more likely that it will prove toxic and you’ll be unable to move forward into clinical trials.
In an incredible stroke of good fortune, our grant proposal to the NIH URGenT program was ultimately funded. This gave us a path forward to do all the remaining studies needed to get to an IND. Through the URGenT network, we will likely ultimately be able to circle back and do the chronic toxicology studies that would make it possible to dose patients for longer, though this remains in the future as of this writing.
For 2.5 years after deciding we would do this ourselves, my to do list was divided in halves. Tasks for the IND, and everything else. I set myself the rule that each day, I had to do everything rate-limiting that was in my court for the IND before I could touch anything else on my to do list. This meant 5:00am inbox sweeps followed by 7:00am Microsoft Teams meetings over breakfast with screaming children in the background, races to mow down piles of DocuSign requests before lab meeting, last-minute calls with consultants to strategize the negotiation of same-day-urgent change orders for yet another need or requirement that we had failed to fully anticipate at the time of original contract signing. And I wasn’t even the front line: my project manager, Alissa Coffey, was the one who solicited and compared all the quotes, reviewed all the contracts, and oversaw all the timelines, a continuously landsliding mountain of work.
By early 2025, all the IND-enabling studies were done, and what remained was to write the IND itself. This, at last, appeared to be the caffeine-drenched 26th mile that this race had been about. Thanks to the URGenT network, we had consultants, but what they wrote, I reviewed, and what they didn’t write, I wrote. Finding templates and examples to work from was a significant challenge. Industry does their INDs to an industrial standard, which is even more than is required for an investigator-initiated IND. Academics who’ve filed INDs, and there aren’t too many examples to begin with, have usually not done it for novel drug modalities, which divalent siRNA is. And INDs, whether industrial or academic, that you can lay eyes on, are very hard to come by. But very generously, a few folks shared their INDs with me, so I had some kind of starting point to work from.
On February 5, 2025, Alissa and I sat together in my office and clicked submit.
what our open IND means

Above: an actual vial of the divalent siRNA drug candidate.
When FDA clears an IND, it doesn’t just clear a drug, it clears a specific batch of drug for a specific patient population, treated on a specific clinical trial protocol. Every detail has to be specified. In our IND, we submitted two trial protocols: one to treat symptomatic patients already diagnosed with prion disease, and one to treat pre-symptomatic patients at risk for prion disease. On March 14, FDA replied that we “may proceed” in symptomatic patients, while we are on “partial clinical hold” for pre-symptomatic patients. That means they want to see more data before they’ll let us dose pre-symptomatic people, and they’ve sent a detailed letter describing what all we’ll need to do to unlock pre-symptomatic dosing. But in the meantime, we have an open IND: we are permitted by FDA to initiate a clinical trial in symptomatic patients with prion disease.
I wish that meant we could start dosing people tomorrow, but the reality is more complicated.
First, we don’t yet have the funding to actually run a clinical trial. NIH has a program for this too, called NeuroNEXT. We worked closely with NIH for months leading up to our IND submission to line everything up, successfully clearing the Stage 1 screening step for this funding mechanism, and within days of hearing back from FDA, we submitted our Stage 2 proposal. Whether this will be funded, and how soon, remains an unknown. The earliest it could possibly happen is a couple of months from now. But it could also be several months, or never.
Suppose that someone wrote us a check today for the whole cost of running a trial. That would be amazing. It would still take time to be up and running. A clinical trial is an extremely complex undertaking. We still need to go through ethical review by the Institutional Review Board (IRB) at one or more hospitals that will serve as trial sites. We haven’t been able to frontload that step, because IRB wants to see that you have funding in place before they’ll agree to review a protocol. Moreover, whether NIH funds the trial will determine several details of how the trial is run and therefore what the IRB needs to review, as the NIH has its own specific standards and expectations for safety monitoring, data entry, statistics, and myriad other aspects of a trial. Once ethical approval is obtained, there will still remain a long list of startup activities, from training staff to initiating databases to finalizing manuals to distributing drug to booking hospital beds.
I’m more keen than anyone to know the timeline of when we’ll start dosing people, but there are genuine unknowns at this point, including that we can’t yet rule out that the answer will be “never,” if we can’t find the funding. When I’ve talked to people experienced in first-in-human clinical trials, most have told me that if funding comes soon, we might plausibly dose a first patient near the end of 2025.
what to expect if and when we are ready to dose patients
If funding enables, at some point in the coming months we may announce that a clinical trial is open and accepting patients. Here are a few of the things you’ll need to keep in mind if and when that happens.
First, a clinical trial is research, not treatment. We are doing a clinical trial precisely because we don’t yet know whether the drug is safe or effective. All drugs do harm to some degree, and most drugs that enter trials never do anyone any good. There are, by definition, not yet any human data to suggest that divalent siRNA will benefit anyone with prion disease. Anyone who volunteers for a trial — this or any trial — should do so on the expectation of contributing to research, not of personally benefitting.
Second, we’re currently limited to giving a single dose of drug. This is because to date, we only have single dose toxicology data in animals. We are working on launching chronic toxicology studies, but we don’t yet know the timeline nor outcome. So at this time we cannot at all promise anyone in the trial that there will ever be a chance to get another dose.
Third, we’re not a commercial sponsor, and while we’re lucky to have been able to come this far, it’s very unlikely we’ll be able to find funding as an academic lab to do the kind of larger clinical trials that would ultimately lead to a drug approval. So we cannot promise a potential future in which this drug is widely and indefinitely available.
Fourth, again due to limited resources, we have not yet explored the possibility of dosing patients outside the United States. We would love for divalent siRNA to ultimately have a global future for patients everywhere. For today, the FDA is the regulatory agency that we know best and that we have the best access to as Americans, so that’s where we went to discuss how to get clearance for a first human dose. Meanwhile, the NIH funding that got us this far was oriented around satisfying U.S. regulatory requirements, and the NIH funding we’ve applied for would only support a trial in the U.S.
For all the above points, remember that it’s not that we don’t want to do more. Our choice so far has been between doing nothing and doing something, and at each juncture, we’ve chosen to do something.
Now let’s talk about what we think we can achieve by doing a clinical trial of divalent siRNA.
The main goal of a trial will be to gain preliminary data on whether the drug is safe at the doses tested. Secondarily, the trial will ask whether the drug was able to engage its target, meaning, lower PrP in the brain, at those same doses. No divalent siRNA for any disease has ever been in humans before. And only 1 drug designed to lower PrP, that is, Ionis’s ION717 ASO, has ever been in humans before. We believe that a small first trial of divalent siRNA in prion disease has the potential to teach us about how divalent siRNA works in the human body — its safety, potency, duration of action — as well as about PrP lowering in prion disease patients.
At the same time, a trial will also teach us about prion disease and how to run clinical trials in this disease. For instance, what kind of patients can we recruit, at what disease stage, how rapidly are they progressing on various measures, and perhaps, what are the kinetics of lowering PrP in this population. We learn a lot from every trial that is run in our disease, and we have much more to learn. The academic-led randomized trials of quinacrine at UCSF 2005-2008 [Geschwind 2013] and of doxycycline in France and Italy 2007-2012 [Haik & Marcon 2014] gave us our first hint of how long patients survive after trial enrollment, but those results are also in desperate need of an update. Prion disease was a different beast back then: both trials took place before CSF RT-QuIC transformed the diagnosis of prion disease, and we hope that we can now find patients at an earlier disease stage. The open label treatment of 6 patients with the PRN100 antibody in the U.K. [Mead 2022] demonstrated the possibility of using MRC Score as an assessment of how advanced the patients were at enrollment into a trial, suggesting the possibility that this could be used as an inclusion criterion to select patients still early enough in their disease. Ionis’s global PrProfile trial of ION717, even while it is still ongoing, has taught us that our patient community is large and motivated: people showed up in such numbers that the trial had to be paused for 4 months. There are tons more lessons to be learned from trials in prion disease, that will be incredibly valuable to the next person designing a trial of (hopefully) an even better drug. Learning some of those lessons in the context of an investigator-initiated study is especially valuable, because we, as academics and especially as patient-scientists, can choose to share data to the greatest degree compatible with patient privacy, in order to maximize generalizeable knowledge and benefit to our whole research community. To be sure, we the patient community can and should pressure companies to share more data publicly, and to some extent they will listen, but publicly traded companies are also under a legal obligation to do right by their shareholders, and realistically, they are never going to share quite as much as we want.
Finally, while we can’t crystal ball what it will look like just yet, it’s possible that divalent siRNA could have a future in prion disease, if the data from a first-in-human trial look good. Good mouse data are a dime a dozen. If it turns out that the human data from a first trial look really promising, that is what would convince a sponsor, someone with more resources than we can muster, to take it on and try to make a drug out of it. Remember, the ultimate goal of our whole quest is a safe and effective drug to treat and prevent prion disease.
what happens if two trials are enrolling at once?
Right now, Ionis’s PrProfile trial has closed enrollment, and Ionis has not yet announced a next trial of ION717. But it is possible that there may be overlap between a future ION717 trial and a future divalent siRNA trial.
Why do we need more than one drug in clinical development? Because we want to maximize the chance of ultimately getting one or more safe and effective drugs. We also want to maximize the research learnings that we gain along the way. Most drugs fail: by various estimates, only 8-14% of drugs that enter a Phase I trial ultimately get approved [Hay 2014, Wong 2019, Thomas 2021]. Even when drugs succeed, it’s rare that the first drug ever in a previously untreatable disease is the one-and-only drug that anyone ever needs. It usually takes many different efforts to finally make a uniformly fatal disease into a manageable or preventable condition. We need multiple shots on goal.
Is it ok to have more than one drug in clinical development? Yes. There are 500 patients diagnosed with prion disease every year in the United States alone. Probably low thousands of cases are diagnosed each year worldwide. Ionis’s PrProfile trial only enrolled 56 patients across the world. The spectacularly fast enrollment of that trial proves that the patients are out there, and are highly motivated. Prion disease is rare, but not ultra-rare. Unfortunately, we have more than enough patients to enroll trials of multiple drugs at the same time. And unfortunately, most patients die very quickly, which means that there is no sense in which enrolling patients this year “uses up” patients for next year.
If patients are eligible for more than one trial, how will they choose which one to enroll in? That’s a conversation with your doctor; I can’t advise you on it. I’ve already shared above some of what I think are the biggest caveats about our drug. If and when a trial is ultimately enrolling, there will be a consent form that will give you a lot more to think about. Above all, remember this is research. As of this writing, there are no human data to suggest that any drug is effective against prion disease.
transparency
As patient-scientists, we want to maximize research on prion disease, and that means a commitment to sharing our learnings and data to the greatest degree feasible. If you’ve followed this blog you know we’ve been big on this for years, but since we’re now at a new stage where human trials could be near at hand, here’s a fresh downpayment on that commitment.
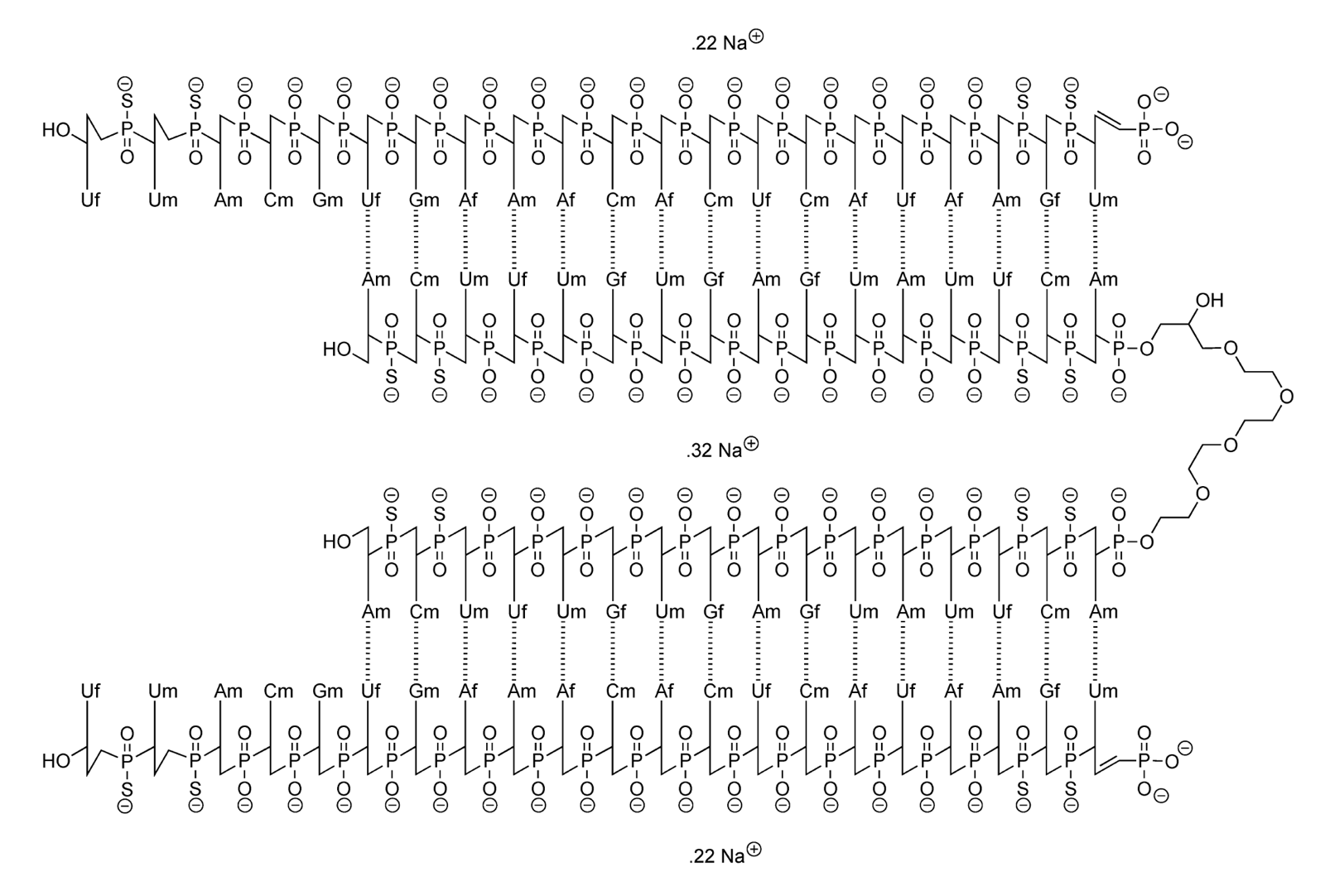
Above: the molecular structure of the drug candidate.
First, we posted a pre-print describing all the preclinical studies we did on divalent siRNA [Gentile 2024]. That includes the exact structure of our drug molecule, something which pharmaceutical companies almost never disclose publicly at an IND or even Phase I clinical trial stage. In fact, there it is again right above this paragraph. The public git repository for the study also includes all the individual-level animal data, something that even most academics never let see the light of day.
Second, we made our IND filing public. Companies don’t do this. Even academics don’t do this. To my knowledge, we are only the third IND ever for which documents have been released publicly (the other two, kudos to them, are an intrathecal AAV gene therapy of AP4M1 for SPG50, and an intrathecal ASO against TNPO2. The more the merrier: I wish 10 or 100 other people would post theirs online. We all have a lot to learn from each other. We posted this IND hoping that if anyone else out there is developing a prion disease drug, it will be a useful starting point to see what we did, and more broadly for the rare disease community, that this might be a useful reference for developing your own drug in your own disease. But remember, it’s just a starting point. Every drug, every drug program, and every indication or disease, is unique. FDA is under no obligation to agree to something for your program just because they said it was okay in our program.
conclusions
We developed a new drug candidate for prion disease in our lab, got permission from FDA to dose human patients, and are now seeking the funding to launch a first-in-human clinical trial. The trial has not launched yet. Don’t write to me asking how you can get the drug: there’s no way just yet. We’re working hard on this. If you can help us find the funding, let me know. When we do get to a trial, it will still be research, not treatment, and no one should go into it on the assumption that it will help them.
If you want to keep apprised of new developments, then follow this blog, and while you’re at it, join our mailing list, and consider joining PrionRegistry if you want to volunteer for research.
Stay tuned.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.