Biomarker changes before disease onset in our MGH cohort study
We’ve just posted a pre-print of our latest findings from a second data freeze of our biomarker cohort study at Mass General [Vallabh 2023]. This post will briefly unpack the context and what we learned.
background
6 years ago, we announced that we were launching a clinical research study at Mass General Hospital with our collaborator, neurologist Steven E. Arnold. The goal was to characterize fluid biomarkers — molecular measurements in blood and cerebrospinal fluid (CSF) — in people at risk for genetic prion disease, plus controls. A lot of research had already been done on biomarkers in symptomatic patients, but at the time, very little was known about what changes might precede onset. In slower-progressing neurodegenerative diseases like Alzheimer’s and Huntington’s, prodromal changes can go on for decades, and many neurologists assumed the same would be true in prion disease.
We put out the call for volunteers (which remains active today, by the way) were thrilled to see that our patient community responded in force. Thank you to everyone who showed up! Within a little over a week, we had more inquiries than we had capacity to enroll people, and thus began the parallel work of brining everyone in, and hunting for funding to expand the study. Luckily, we’ve been successful, and with support from Prion Alliance, CJD Foundation, a federal grant from NCATS, and Ionis Pharmaceuticals, the study has now grown to 62 people. It’s remarkable to see that year after year, healthy people use their paid vacation days off of work to fly to Boston and undergo lumbar punctures with no prospect of personally benefitting from it, just to advance research. One side effect of this study is that it has demonstrated just how motivated our patient community is, and has smashed a lot of doubts about whether we can find the pre-symptomatic patients, or whether they’ll be willing to undergo lumbar punctures.
what we knew up to now
In the first data freeze, published in 2020, we got to tell a mostly happy story [Vallabh 2020a]. First, CSF PrP was stable over time, at least in the short term, just as we had hoped. The mean coefficient of variation in one person over time was just 7%, which suggested that if a drug could lower PrP in the brain, it would be really easy to read that out in a trial by measuring PrP in CSF. Second, asymptomatic mutation carriers were generally negative for biomarker signs of disease. We did not see elevated NfL or T-Tau — two markers of neuronal damage that go way up in symptomatic patients.
But in 1 out of 23 people, we did see RT-QuIC positivity in CSF. At the time, we wrote that the prognostic value was unknown. In patients who present with rapidly progressive dementia, CSF RT-QuIC is highly sensitive and exquisitely specific, and has become the diagnostic test for diagnosing prion disease. RT-QuIC works really well in sporadic prion disease, and in E200K, but not so well for other genetic mutations, where it is usually negative even after symptom onset. The 1 positive individual in our study was E200K.
We’re thankfully not the only ones studying biomarkers in pre-symptomatic people, and in a report from London earlier this year, T.H. Mok and Simon Mead found 3 more pre-symptomatic E200K individuals with positive CSF RT-QuIC. One developed disease just a few months later, but the other two remained healthy as of writing. Both that report [Mok 2023] and a prior one from London [Thompson 2021] also described changes in other markers, such as NfL (measuring neuronal damage) and GFAP (measuring neuroinflammation) prior to onset. A caveat is that, as I argued in 2020, just because NfL rises in people who develop disease doesn’t necessarily mean it would be easy, prospectively, to predict who will develop disease by looking at NfL. NfL varies naturally from person to person and rises over time even in normal controls, so interpreting it probably requires some kind of normalization both to individual baseline and to age, and even then, the sensitivity and specificity of it for oncoming disease onset may be limited.
the new findings
The new pre-print describes our second data freeze, capturing about 5.5 years of follow-up on our cohort. It encompasses 62 people — 41 PRNP mutation carriers and 21 controls — well matched in terms of age and sex, with a total of >150 CSF and plasma samples collected.
Here comes the not-so-happy part of the story: 4 participants in the cohort have now gone on to develop active disease. When we started this study, it felt like trials of a PrP-lowering drug might be just around the corner, and that pre-symptomatic people would be eligible. We hoped that soon we’d be able to go back to our research volunteers and ask if they wanted to enroll in a drug trial. That is still our goal, and I remain a fervent optimist for the long term. But with one delay after another, the timeline has not been what we once hoped. As you know if you’ve followed this blog, only earlier this month has the first trial of a PrP-lowering ASO been posted, and it’s still not recruiting yet, but when it does, it will be limited to symptomatic patients. In the years that we’ve been working to make trials a reality, hope hasn’t come soon enough for everyone. While I wish we could have given them the gift of even an experimental drug, even a chance at something helpful, instead these 4 participants have given us a rare and precious gift: the opportunity to observe biomarker changes leading up to disease onset.
We sum up all the findings in one big figure:
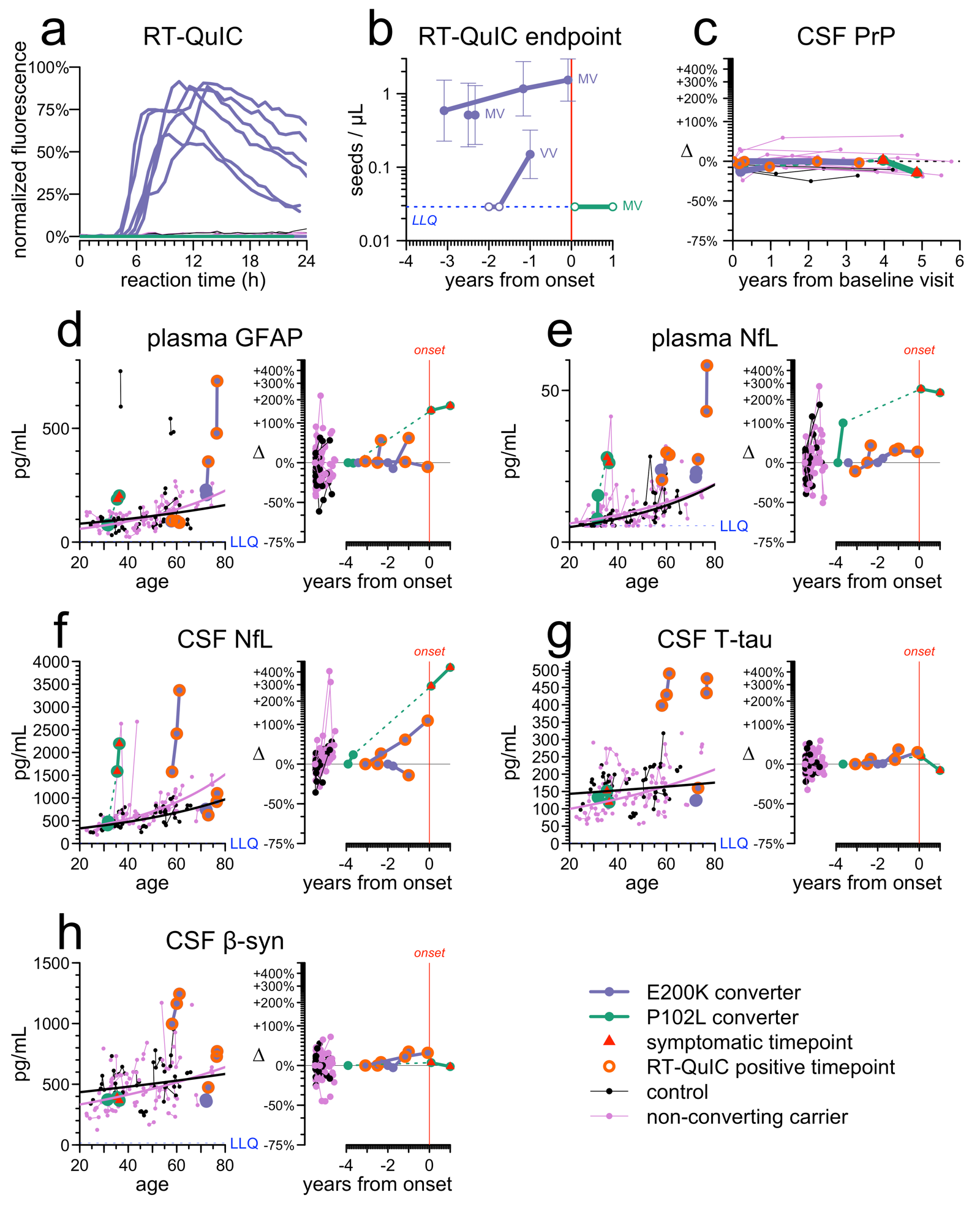
Out of 149 CSF samples from our cohort tested by RT-QuIC, we observed 6 positives, all of which belonged to 3 E200K individuals who went on to develop disease. While it is still early days and small sample numbers, this gives us some hint that RT-QuIC may indeed be prognostic. We also suspect it may be genotype-dependent. 2 individuals who were heterozygous (MV) at PRNP codon 129 were already positive at first sample ~3 years before onset, while 1 individual who was homozygous (VV) started out negative and then became positive just 1 year before onset. That gives us a hint that the duration of RT-QuIC positivity may depend on codon 129 genotype. That would make sense, since we know that disease duration after onset is longer for MV individuals, and durations of prodrome and disease appear correlated. At T.H. Mok’s suggestion, we did endpoint dilution on the CSF to determine the titer, or concentration, of prion seeds, but the titer did not appear to rise measurably over time — whenever it was positive, it was a strong positive.
We know that CSF PrP goes down in symptomatic disease, probably because PrP is caught up in plaques in the brain instead of being shed into CSF. But it doesn’t seem to change pre-symptomatically. CSF PrP is lower in people with mutations, but that seems to be a constitutive lifelong change, rather than a disease-related prodromal change, because pre-symptomatic people remain remarkably steady in CSF PrP concentration, with a mean coefficient of variation of 10% even over years. Interestingly, this appears true even in people who become RT-QuIC positive. Their CSF PrP concentration was incredibly steady right up to onset.
We also looked at 5 other biomarkers of neuronal damage and neuroinflammation. For this, after much debate, we devised a special data visualization with side-by-side plots for each. The left panel shows the biomarker value relative to age, so you can see whether a person was anomalous relative to the overall increase with age in normal individuals. The right panel shows the biomarker value normalized to individual baseline and plotted against years of follow-up, so you can see whether the fold chnage was remarkable or not.
Plasma NfL was perhaps the most promising, in that it’s both high and increasing in all 4 individuals. But even still, if we’d had to predict onsets based on plasma NfL, we’d have been wrong more often than right — the highest age-normalized values and the highest fold changes did not always belong to the people who converted. All the other markers contributed something as well, but were even less able to stand alone. For each of CSF NfL, plasma GFAP, CSF T-tau, and CSF beta-synuclein, 2/4 people who developed disease were elevated, but it was a different 2/4 each time.
take-home conclusions
This is just 4 disease onsets and so still a very small number on which to base conclusions, but this being a rare disease, we have to learn what we can. It appears that RT-QuIC may be positive for 1-3 years before onset in E200K individuals, and that the duration of positivity could be correlated with codon 129 genotype. Neuronal damage and neuroinflammation markers could play a supporting role in identifying people proximal to onset, but are unlikely to be sufficient on their own to reliably predict who will develop disease.
There are two calls to action here. First, we must continue to expand pre-symptomatic cohorts both cross-sectionally and longitudinally. We are eager to hear future dispatches from other groups studying pre-symptomatic people, including UCSF, Tel Aviv Medical Center [Bregman 2023], Raquel Sanchez in Barcelona, Inga Zerr in Germany, and so on. More numbers will be critical to determining prognostic value of all these markers. Second, we need to develop new candidate prodromal markers for other genetic subtypes, since RT-QuIC is really only sensitive to E200K.
In mice, there is an outsize benefit to treating early [Minikel 2020], and we continue to believe that our best chance at prevention is primary prevention — recruiting based on genetic risk only, and treating to a CSF PrP endpoint [Vallabh 2020b]. Moreover, there aren’t enough prodromal people, cross-sectionally, to power an entire trial. But there may be times when an experimental drug just does not yet have the safety profile to go into completely healthy, biomarker-negative at-risk people, where perhaps justifying the first pre-symptomatic test would require showing that someone is very likely close to onset. Or in a trial of pre-symptomatic people, perhaps there could be a compassionate use off-ramp for people who develop signs of imminent disease. While prodromal changes shouldn’t be our only hope for preventing prion disease, they could play a supportive role, and we have much work ahead of us to better establish these markers.
We owe an enormous debt of gratitude to all our research participants, but especially those for whom we didn’t get there fast enough. It is our duty to do justice by the gift you’ve given us, by using it to find ways to prevent this horrible disease before it takes more of us.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.